

Requirement of cyclin E-Cdk2 inhibition in p16(INK4a)-mediated growth suppression
- PMID: 9710613
- PMCID: PMC109114
- DOI: 10.1128/MCB.18.9.5284
Requirement of cyclin E-Cdk2 inhibition in p16(INK4a)-mediated growth suppression
Abstract
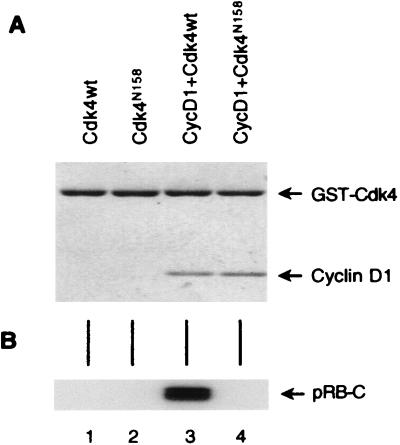
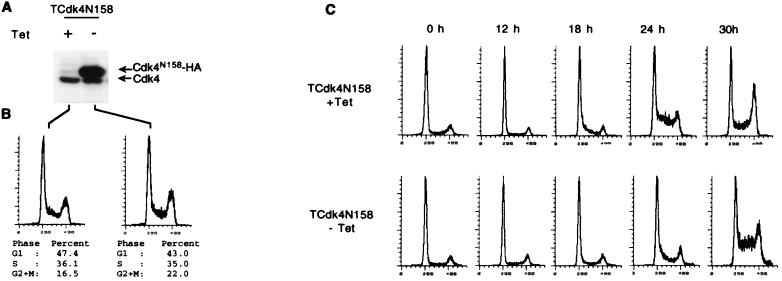
Loss-of-function mutations of p16(INK4a) have been identified in a large number of human tumors. An established biochemical function of p16 is its ability to specifically inhibit cyclin D-dependent kinases in vitro, and this inhibition is believed to be the cause of the p16-mediated G1 cell cycle arrest after reintroduction of p16 into p16-deficient tumor cells. However, a mutant of Cdk4, Cdk4(N158), designed to specifically inhibit cyclin D-dependent kinases through dominant negative interference, was unable to arrest the cell cycle of the same cells (S. van den Heuvel and E. Harlow, Science 262:2050-2054, 1993). In this study, we determined functional differences between p16 and Cdk4(N158). We show that p16 and Cdk4(N158) inhibit the kinase activity of cellular cyclin D1 complexes through different mechanisms. p16 dissociated cyclin D1-Cdk4 complexes with the release of bound p27(KIP1), while Cdk4(N158) formed complexes with cyclin D1 and p27. In cells induced to overexpress p16, a higher portion of cellular p27 formed complexes with cyclin E-Cdk2, and Cdk2-associated kinase activities were correspondingly inhibited. Cells engineered to express moderately elevated levels of cyclin E became resistant to p16-mediated growth suppression. These results demonstrate that inhibition of cyclin D-dependent kinase activity may not be sufficient to cause G1 arrest in actively proliferating tumor cells. Inhibition of cyclin E-dependent kinases is required in p16-mediated growth suppression.
Figures





References
-
- Baldin V, Lukas J, Marcote M J, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 1993;7:812–821. - PubMed
-
- Bates S, Parry D, Bonetta L, Vousden K, Dickson C, Peters G. Absence of cyclin D/cdk complexes in cells lacking functional retinoblastoma. Oncogene. 1994;9:1633–1640. - PubMed
-
- Blain S W, Montalvo E, Massague J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem. 1996;272:25863–25872. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous