

Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice
- PMID: 9724715
- PMCID: PMC27906
- DOI: 10.1073/pnas.95.18.10402
Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice
Abstract
ATP-sensitive K+ (KATP) channels regulate many cellular functions by linking cell metabolism to membrane potential. We have generated KATP channel-deficient mice by genetic disruption of Kir6.2, which forms the K+ ion-selective pore of the channel. The homozygous mice (Kir6.2(-/-)) lack KATP channel activity. Although the resting membrane potential and basal intracellular calcium concentrations ([Ca2+]i) of pancreatic beta cells in Kir6.2(-/-) are significantly higher than those in control mice (Kir6.2(+/+)), neither glucose at high concentrations nor the sulfonylurea tolbutamide elicits a rise in [Ca2+]i, and no significant insulin secretion in response to either glucose or tolbutamide is found in Kir6.2(-/-), as assessed by perifusion and batch incubation of pancreatic islets. Despite the defect in glucose-induced insulin secretion, Kir6.2(-/-) show only mild impairment in glucose tolerance. The glucose-lowering effect of insulin, as assessed by an insulin tolerance test, is increased significantly in Kir6.2(-/-), which could protect Kir6.2(-/-) from developing hyperglycemia. Our data indicate that the KATP channel in pancreatic beta cells is a key regulator of both glucose- and sulfonylurea-induced insulin secretion and suggest also that the KATP channel in skeletal muscle might be involved in insulin action.
Figures
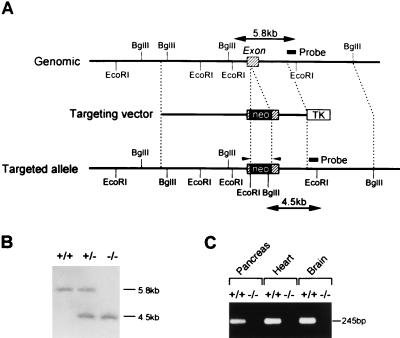
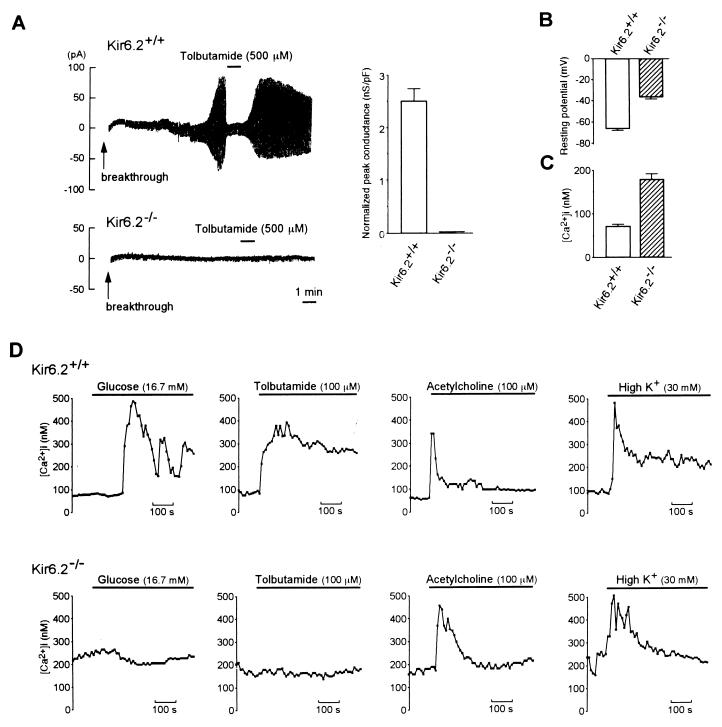
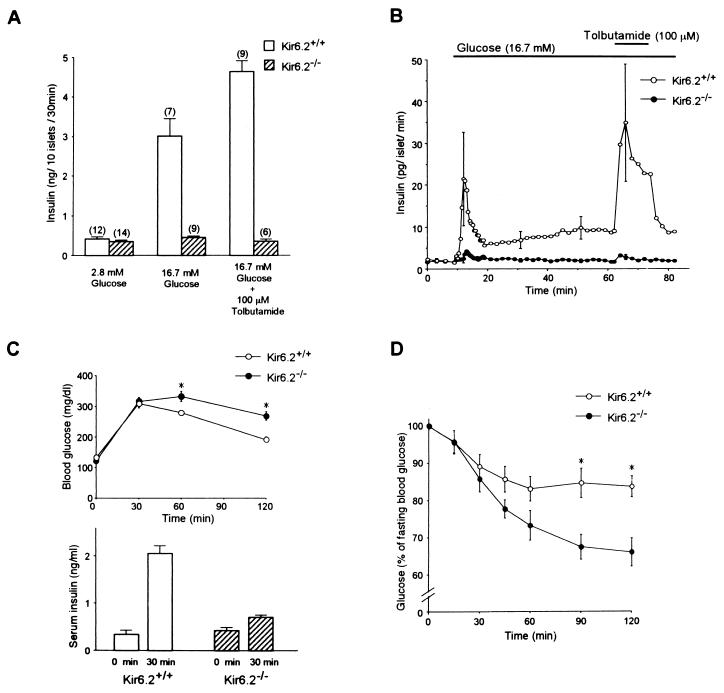
References
-
- Noma A. Nature (London) 1983;305:147–148. - PubMed
-
- Cook D L, Hales C N. Nature (London) 1984;310:271–273. - PubMed
-
- Ashcroft F M, Harrison D E, Ashcroft S J H. Nature (London) 1984;312:446–448. - PubMed
-
- Spruce A E, Standen N B, Stanfield P R. Nature (London) 1985;316:736–738. - PubMed
-
- Standen N B, Quayle J M, Davies N W, Brayden J E, Huang Y, Nelson M T. Science. 1989;245:177–180. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
