

doi: 10.1128/JVI.72.10.8396-8402.1998.
Specific and independent recognition of U3 and U5 att sites by human immunodeficiency virus type 1 integrase in vivo
Affiliations
- PMID: 9733892
- PMCID: PMC110226
- DOI: 10.1128/JVI.72.10.8396-8402.1998
Item in Clipboard
Specific and independent recognition of U3 and U5 att sites by human immunodeficiency virus type 1 integrase in vivo
J Virol.
1998 Oct.
Abstract
The retroviral attachment (att) sites at viral DNA ends are cis-acting regions essential for proviral integration. To investigate the sequence features of att important for human immunodeficiency virus type 1 (HIV-1) integration in vivo, we generated a series of 25 att mutants of HIV-1 by mutagenesis of the U3, U5, or both boundaries of att. Our results indicated that the terminal 11 or 12 bp of viral DNA are sufficient for specific recognition by HIV-1 integrase (IN) and suggested that IN might recognize each att site independently in vivo.
Figures

Mutation of HIV-1 U3 and U5 att sites. The boundary and nucleotide sequences of the U5 (A) and U3 (B) att sites are shown. Both strands of the WT DNA sequences are shown at the top. The highly conserved CA and TG dinucleotides in each att site are underlined. For att site mutants, only positive-strand DNA sequences are shown. A deleted or altered nucleotide is indicated by a dash or a bold letter, respectively. In MuLV or FIV att chimeric mutants, the terminal 13 bp of each HIV-1 att sequence were replaced with the corresponding terminal 13 bp of MuLV or FIV. The boundary of the primer binding site (pbs) for tRNA3Lys is shown by dashed underlining (A).
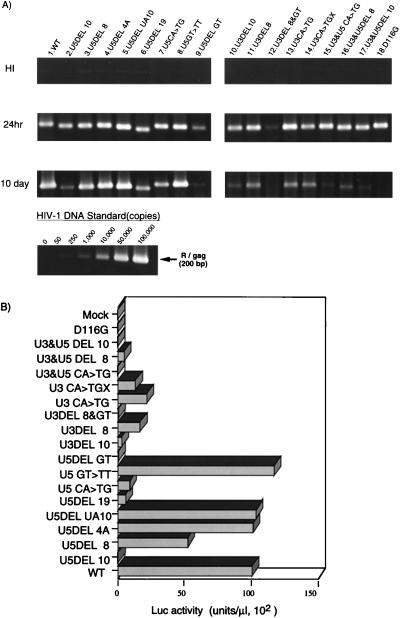
Analysis of att mutants. (A) For each virus, 106 RD cells were infected by inoculating a 1-ml aliquot of DNase-treated, virus-containing COS cell supernatant. After 24 h or 10 days, as indicated on the left, the entire cell culture was harvested. Total DNA was extracted from infected RD cells and subjected to quantitative PCR analysis with primer pairs specific for the R/gag region of HIV-1 (38, 58). For HIV-1 DNA standards, 50 to 100,000 copies of linearized HIVNLluc-env were amplified in parallel. Amplified products were resolved on a 2% agar gel and visualized by Sybr Green staining (FMC Bio Product, Rockland, Maine). Quantitative analysis of amplified products was performed by the Epi-Light UV FA1100 system with a Luminous Imager (Aisin Cosmos R&D Co.). An aliquot of each virus preparation was incubated at 65°C for 30 min and used as a heat-inactivated control (HI). (B) At 3 days postinfection, the entire culture was harvested and washed twice with phosphate-buffered saline. The cell pellet was resuspended with 200 μl of cell lysate buffer (Promega Corp.). Ten microliters of each cell lysate was subjected to a Luc assay. Luc activity was determined after subtraction of the background level and normalization to 1 μl of lysate, corresponding to about 103 cells. Luc activity was measured in duplicate for each virus inoculate, and mean values are shown. The standard error for each replicate was less than 10% of the mean. This experiment was performed three times with independently prepared virus. The results of a representative experiment are shown. A virus prepared with a parental (WT) vector or an IN catalytic mutant (D116G) vector was infected in parallel as a WT or integration-defective control, respectively. Mock, mock-transfected COS cells (no virus).
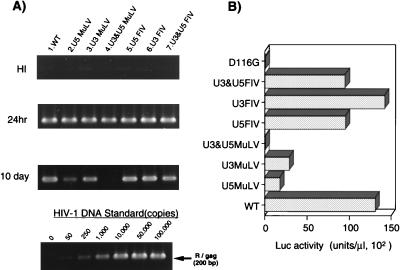
Analysis of att chimeric mutants. A 1-ml aliquot of each virus containing about 50 ng of p24 was inoculated into 106 RD cells. (A) At 24 h or 10 days after infection, as indicated on the left, total DNA was extracted and subjected to quantitative PCR analyses of de novo-synthesized viral DNA as described in the legend to Fig. 2. (B) For each virus, at 3 days postinfection of the same virus preparation used for the PCR experiment in panel A, the entire culture was harvested. The cell pellet fraction was subjected to a Luc assay as described in the legend to Fig. 2. Luc activity was measured in duplicate for each virus inoculate, and mean values are shown. The standard error for each replicate was less than 10% of the mean. A virus prepared with a parental (WT) or an IN catalytic mutant (D116G) vector was infected in parallel as a WT or integration-defective control, respectively. This experiment was performed three times with independently prepared virus. The results of a representative experiment are shown.
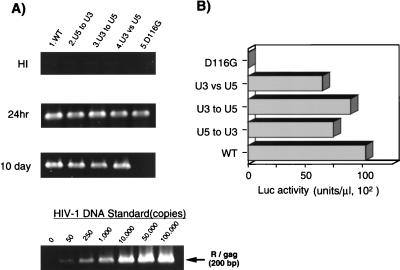
Analysis of att exchange mutants. Each virus was inoculated into 106 RD cells as described in the legend to Fig. 2. (A) At 24 h or 10 days after infection, as indicated on the left, total DNA was extracted and subjected to quantitative PCR analyses of de novo-synthesized viral DNA as described in the legend to Fig. 2. (B) For each virus, at 3 days postinfection with the same virus preparation used for the PCR experiment in panel A, the entire culture was harvested and subjected to a Luc assay as described in the legend to Fig. 2. Luc activity was measured in duplicate for each virus inoculate, and mean values are shown. The standard error for each replicate was less than 10% of the mean. A virus prepared with a parental (WT) vector or an IN catalytic mutant (D116G) vector was infected in parallel as a WT or integration-defective control, respectively. This experiment was performed three times with independently prepared virus. The results of a representative experiment are shown.
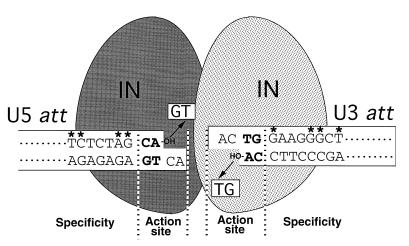
Specific att site recognition by HIV-1 IN. The U3 and U5 att regions at both termini of the viral DNA (shown as open boxes) are recognized and subjected to an integration reaction by IN. Removal of the terminal GT dinucleotide by IN (illustrated with arrows) resulted in exposure of the conserved dinucleotide CA-OH (shown in bold letters) at the 3′ ends of both strands. The U3 (12-bp) and U5 (11-bp) att sequences of HIV-1, which are required for efficient interaction with IN, are shown in the boxes. The nucleotides suggested to be important for specific interaction with HIV-1 IN are indicated by asterisks. The independent recognition of each att site by each IN promoter by dimer formation is speculatively illustrated.
Similar articles
-
Activity of recombinant HIV-1 integrase on mini-HIV DNA.Nucleic Acids Res. 1999 May 15;27(10):2202-10. doi: 10.1093/nar/27.10.2202. Nucleic Acids Res. 1999. PMID: 10219094 Free PMC article.
-
Genetic analysis of human immunodeficiency virus type 1 integrase and the U3 att site: unusual phenotype of mutants in the zinc finger-like domain.J Virol. 1995 Nov;69(11):6687-96. doi: 10.1128/JVI.69.11.6687-6696.1995. J Virol. 1995. PMID: 7474078 Free PMC article.
-
Both substrate and target oligonucleotide sequences affect in vitro integration mediated by human immunodeficiency virus type 1 integrase protein produced in Saccharomyces cerevisiae.J Virol. 1992 Apr;66(4):2359-68. doi: 10.1128/JVI.66.4.2359-2368.1992. J Virol. 1992. PMID: 1548767 Free PMC article.
-
Site-specific integration of retroviral DNA in human cells using fusion proteins consisting of human immunodeficiency virus type 1 integrase and the designed polydactyl zinc-finger protein E2C.Methods. 2009 Apr;47(4):269-76. doi: 10.1016/j.ymeth.2009.01.001. Epub 2009 Jan 30. Methods. 2009. PMID: 19186211 Free PMC article. Review.
-
Integrase and integration: biochemical activities of HIV-1 integrase.Retrovirology. 2008 Dec 17;5:114. doi: 10.1186/1742-4690-5-114. Retrovirology. 2008. PMID: 19091057 Free PMC article. Review.
Cited by
-
Integration-deficient lentiviral vectors: a slow coming of age.Mol Ther. 2009 Aug;17(8):1316-32. doi: 10.1038/mt.2009.122. Epub 2009 Jun 2. Mol Ther. 2009. PMID: 19491821 Free PMC article. Review.
-
Asymmetric processing of human immunodeficiency virus type 1 cDNA in vivo: implications for functional end coupling during the chemical steps of DNA transposition.Mol Cell Biol. 2001 Oct;21(20):6758-67. doi: 10.1128/MCB.21.20.6758-6767.2001. Mol Cell Biol. 2001. PMID: 11564861 Free PMC article.
-
Molecular Interactions between HIV-1 integrase and the two viral DNA ends within the synaptic complex that mediates concerted integration.J Mol Biol. 2009 May 29;389(1):183-98. doi: 10.1016/j.jmb.2009.04.007. Epub 2009 Apr 9. J Mol Biol. 2009. PMID: 19362096 Free PMC article.
-
SiRNA-induced mutation in HIV-1 polypurine tract region and its influence on viral fitness.PLoS One. 2015 Apr 10;10(4):e0122953. doi: 10.1371/journal.pone.0122953. eCollection 2015. PLoS One. 2015. PMID: 25860884 Free PMC article.
-
Mutations in the U5 region adjacent to the primer binding site affect tRNA cleavage by human immunodeficiency virus type 1 reverse transcriptase in vivo.J Virol. 2008 Jan;82(2):719-27. doi: 10.1128/JVI.02611-06. Epub 2007 Nov 7. J Virol. 2008. PMID: 17989171 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
