

Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor
- PMID: 9751783
- PMCID: PMC21758
- DOI: 10.1073/pnas.95.20.12022
Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor
Abstract
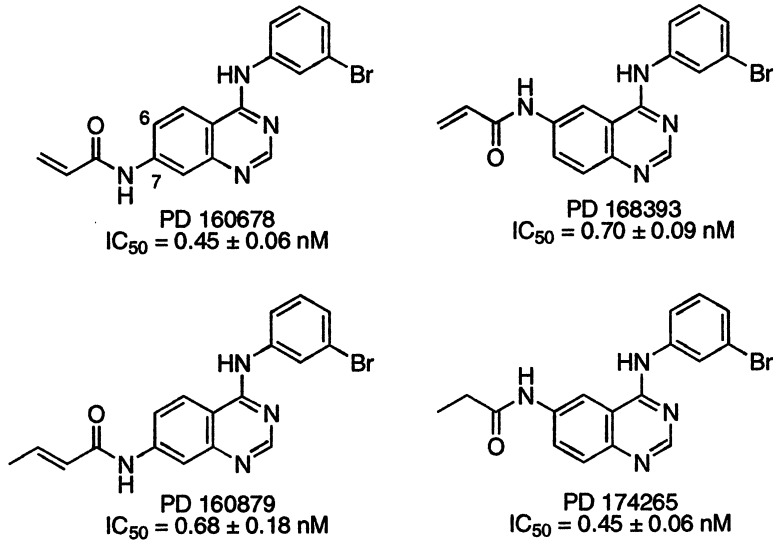
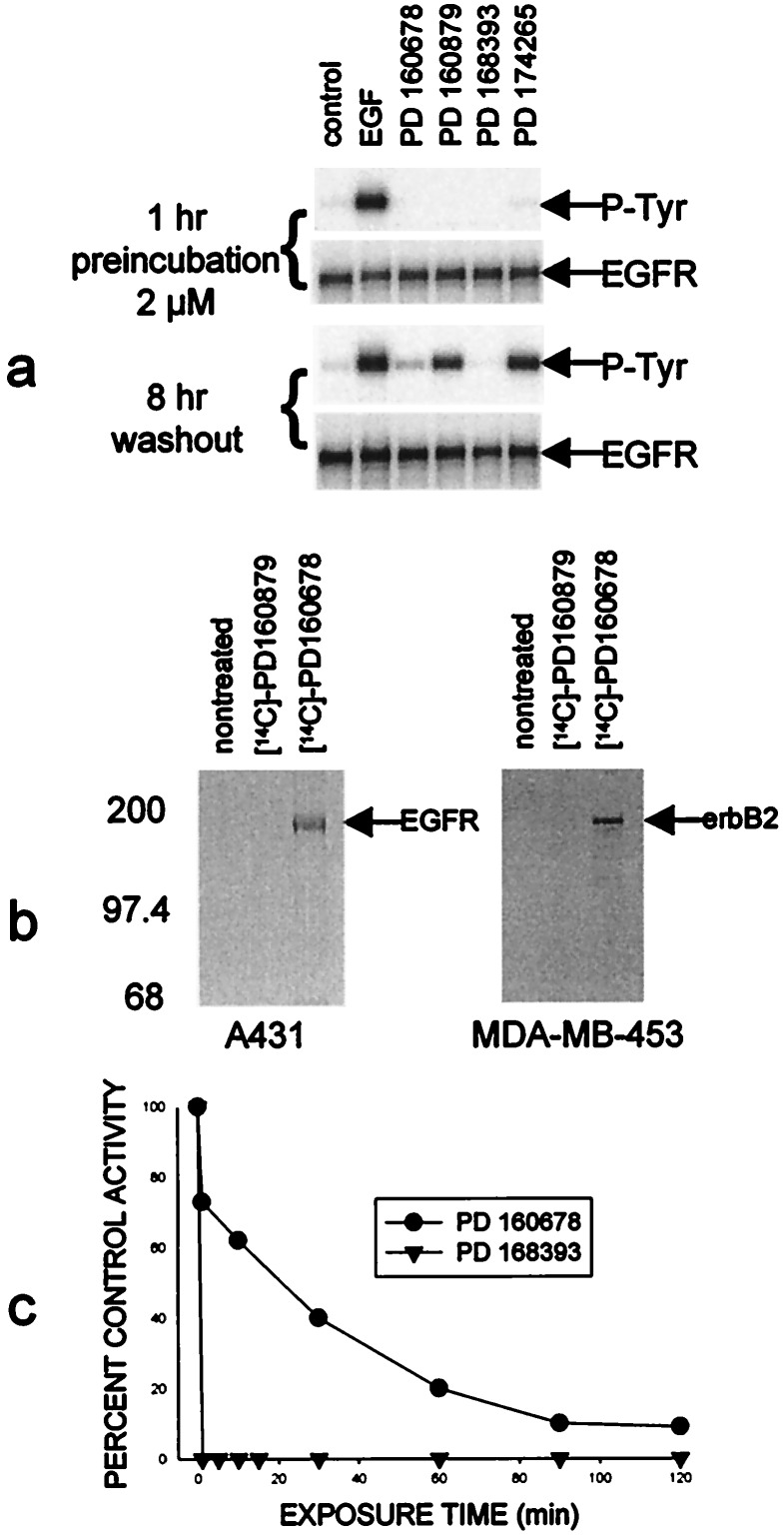
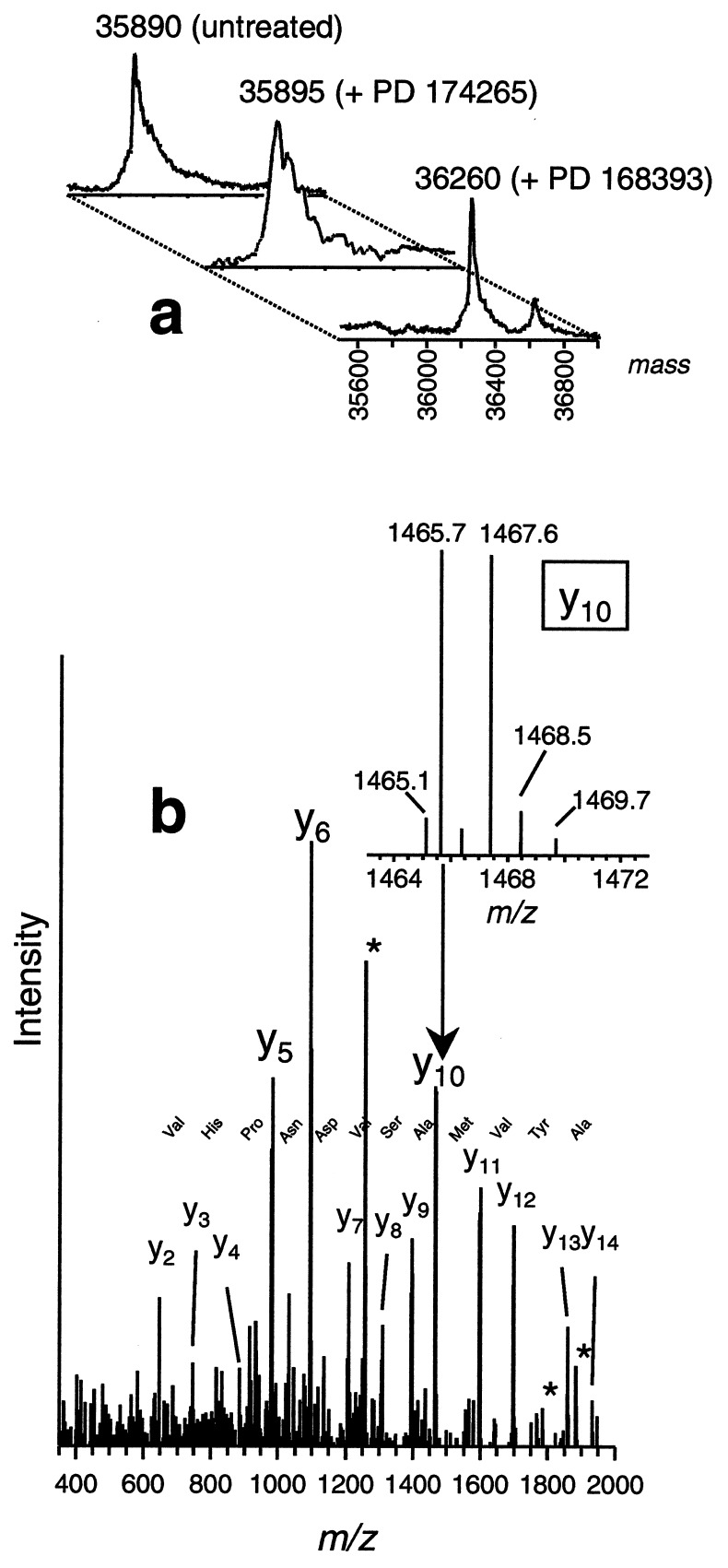
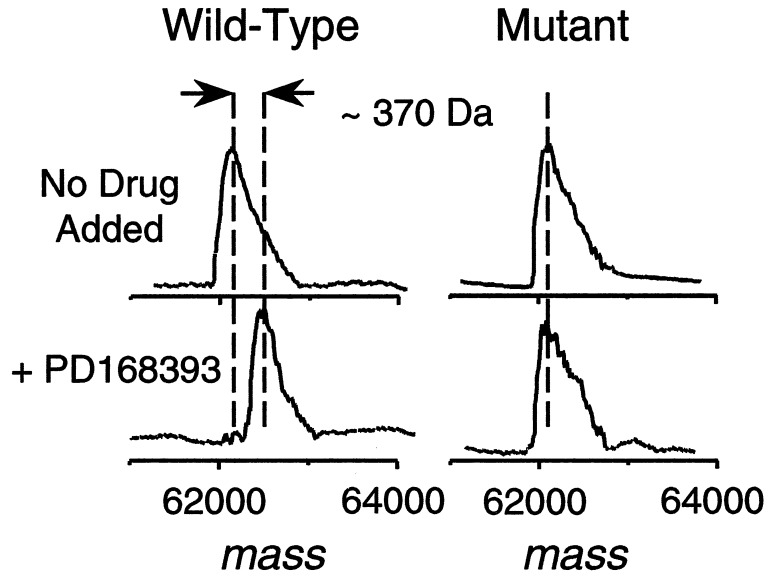
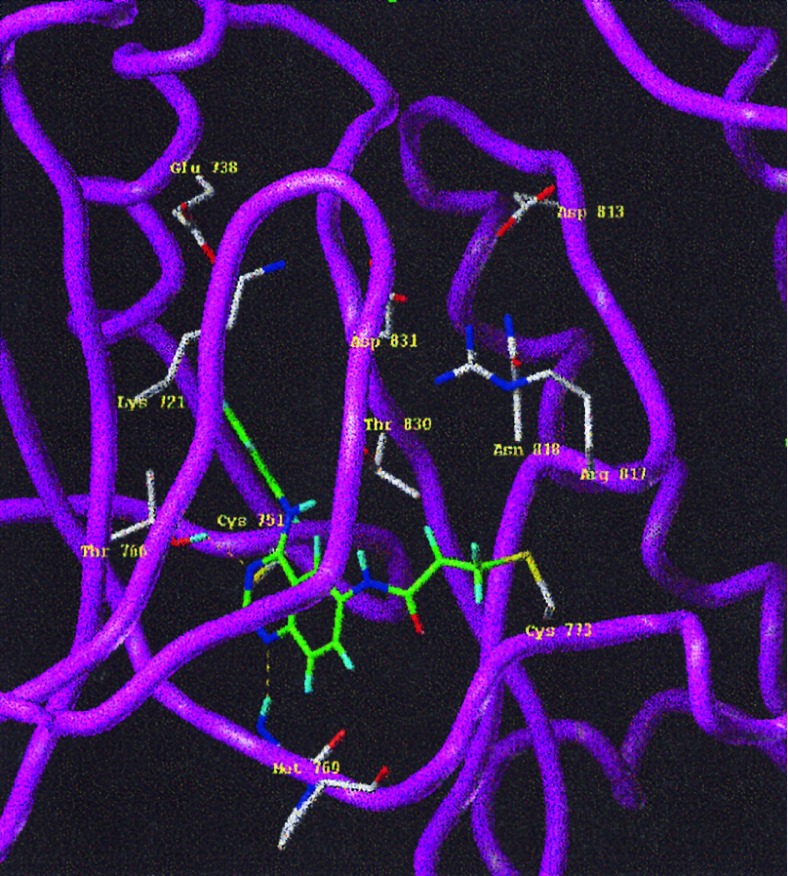
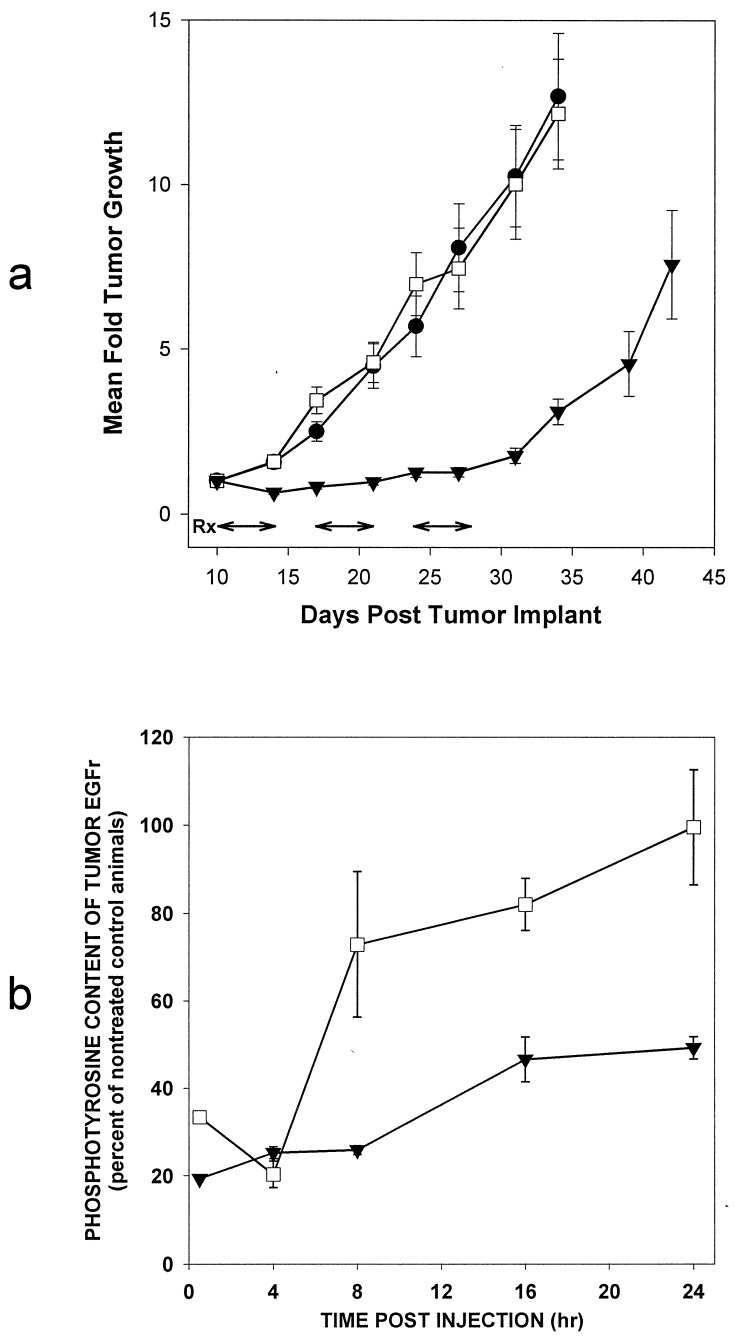
A class of high-affinity inhibitors is disclosed that selectively target and irreversibly inactivate the epidermal growth factor receptor tyrosine kinase through specific, covalent modification of a cysteine residue present in the ATP binding pocket. A series of experiments employing MS, molecular modeling, site-directed mutagenesis, and 14C-labeling studies in viable cells unequivocally demonstrate that these compounds selectively bind to the catalytic domain of the epidermal growth factor receptor with a 1:1 stoichiometry and alkylate Cys-773. While the compounds are essentially nonreactive in solution, they are subject to rapid nucleophilic attack by this particular amino acid when bound in the ATP pocket. The molecular orientation and positioning of the acrylamide group in these inhibitors in relation to Cys-773 entirely support these results as determined from docking experiments in a homology-built molecular model of the ATP site. Evidence is also presented to indicate that the compounds interact in an analogous fashion with erbB2 but have no activity against the other receptor tyrosine kinases or intracellular tyrosine kinases that were tested in this study. Finally, a direct comparison between 6-acrylamido-4-anilinoquinazoline and an equally potent but reversible analog shows that the irreversible inhibitor has far superior in vivo antitumor activity in a human epidermoid carcinoma xenograft model with no overt toxicity at therapeutically active doses. The activity profile for this compound is prototypical of a generation of tyrosine kinase inhibitors with great promise for therapeutic significance in the treatment of proliferative disease.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
