

Molecular mimicry and immune-mediated diseases
- PMID: 9761770
- PMCID: PMC7164021
- DOI: 10.1096/fasebj.12.13.1255
Molecular mimicry and immune-mediated diseases
Abstract
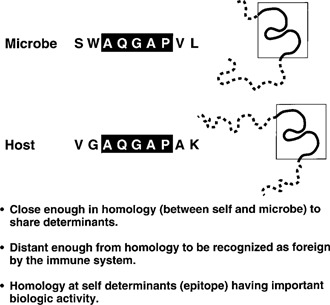
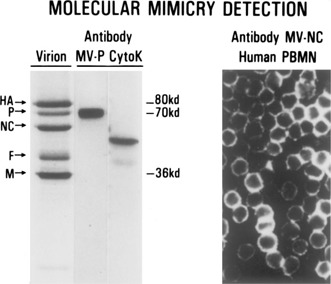
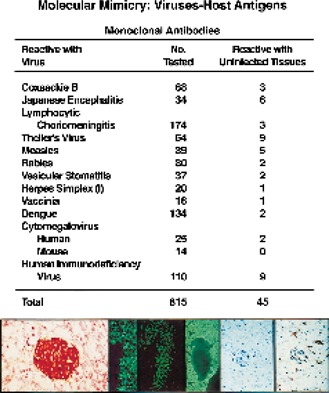
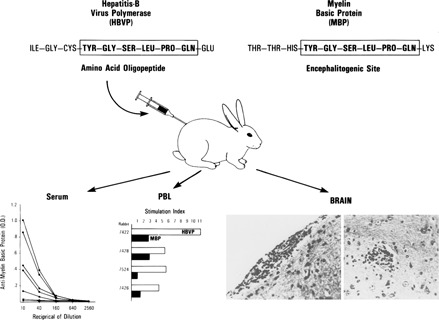
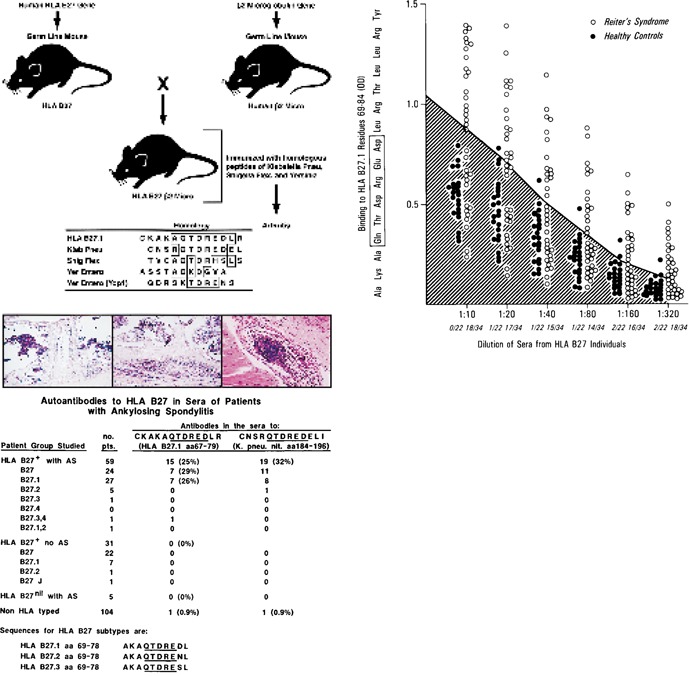
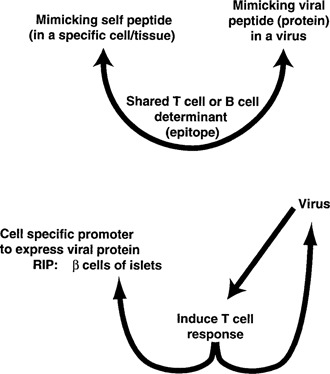
Molecular mimicry has been proposed as a pathogenetic mechanism for autoimmune disease, as well as a probe useful in uncovering its etiologic agents. The hypothesis is based in part on the abundant epidemiological, clinical, and experimental evidence of an association of infectious agents with autoimmune disease and observed cross-reactivity of immune reagents with host 'self' antigens and microbial determinants. For our purpose, molecular mimicry is defined as similar structures shared by molecules from dissimilar genes or by their protein products. Either the molecules' linear amino acid sequences or their conformational fits may be shared, even though their origins are as separate as, for example, a virus and a normal host self determinant. An immune response against the determinant shared by the host and virus can evoke a tissue-specific immune response that is presumably capable of eliciting cell and tissue destruction. The probable mechanism is generation of cytotoxic cross-reactive effector lymphocytes or antibodies that recognize specific determinants on target cells. The induction of cross-reactivity does not require a replicating agent, and immune-mediated injury can occur after the immunogen has been removed a hit-and-run event. Hence, the viral or microbial infection that initiates the autoimmune phenomenon may not be present by the time overt disease develops. By a complementary mechanism, the microbe can induce cellular injury and release self antigens, which generate immune responses that cross-react with additional but genetically distinct self antigens. In both scenarios, analysis of the T cells or antibodies specifically engaged in the autoimmune response and disease provides a fingerprint for uncovering the initiating infectious agent.
Figures
References
-
- Theofilopoulos, A. (1995) The basis of autoimmunity. II. Genetic predisposition. Immunol. Today 16, 150–159 - PubMed
-
- Merriman, T. R. , and Todd, J. A. (1995) Genetics of autoimmune disease. Curr. Opin. Immunol. 7, 786–792 - PubMed
-
- Brewerton, D. , Caffrey, M. , Hart, F. , James, D. , Nichols, A. , and Sturrock, R. (1973) Ankylosing spondylitis and HLA B27. Lancet i, 904–907 - PubMed
-
- Green, A. (1990) The role of genetic factors in development of IDDM. Curr. Top. Microbiol. Immunol. 164, 3–17 - PubMed
-
- Ebers, G. , Bulman, D. , Sadovnik, A. , Paty, D. W. , Warren, S. , Hader, W. , Murray, T. J. , Seland, T. P. , Duquette, P. , Grey, T. , et al. (1987) A population‐based study of multiple sclerosis in twins. New Engl. J. Med. 315, 1638–1642 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
