

Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome
- PMID: 9763493
- PMCID: PMC6792844
- DOI: 10.1523/JNEUROSCI.18-20-08505.1998
Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome
Abstract
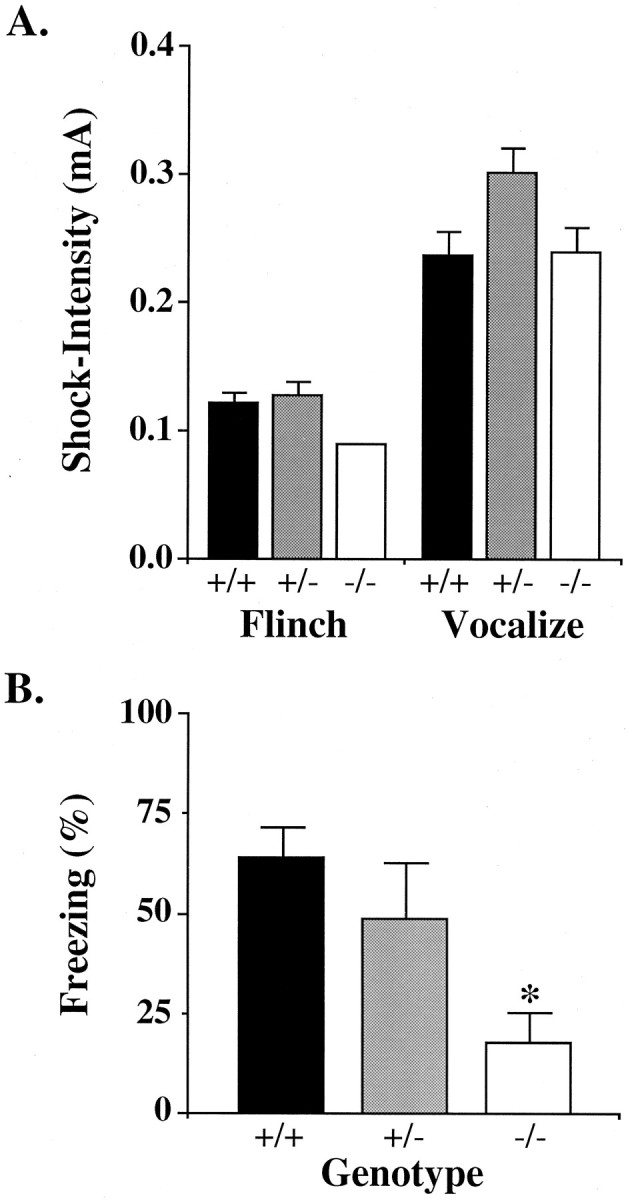
Angelman syndrome (AS) is a severe neurodevelopmental disorder resulting from a deletion/mutation in maternal chromosome 15q11-13. The genes in 15q11-13 contributing to the full array of the clinical phenotype are not fully identified. This study examines whether a loss or reduction in the GABAA receptor beta3 subunit (GABRB3) gene, contained within the AS deletion region, may contribute to the overall severity of AS. Disrupting the gabrb3 gene in mice produces electroencephalographic abnormalities, seizures, and behavior that parallel those seen in AS. The seizures that are observed in these mice showed a pharmacological response profile to antiepileptic medications similar to that observed in AS. Additionally, these mice exhibited learning and memory deficits, poor motor skills on a repetitive task, hyperactivity, and a disturbed rest-activity cycle, features all common to AS. The loss of the single gene, gabrb3, in these mice is sufficient to cause phenotypic traits that have marked similarities to the clinical features of AS, indicating that impaired expression of the GABRB3 gene in humans probably contributes to the overall phenotype of Angelman syndrome. At least one other gene, the E6-associated protein ubiquitin-protein ligase (UBE3A) gene, has been implicated in AS, so the relative contribution of the GABRB3 gene alone or in combination with other genes remains to be established.
Figures







References
-
- Bottani A, Robinson WP, DeLozier-Blanchet CD, Engel E, Morris MA, Schmitt B, Thun-Hohenstein L, Schinzel A. Angelman syndrome due to paternal uniparental disomy of chromosome 15: a milder phenotype? Am J Med Genet. 1994;51:35–40. - PubMed
-
- Boyd SG, Harden A, Patton MA. The EEG in early diagnosis of the Angelman (Happy Puppet) syndrome. Eur J Pediatr. 1988;147:508–513. - PubMed
-
- Buiting K, Saitoh S, Gross S, Dittrich B, Schwartz S, Nicholls RD, Horsthemke B. Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15. Nat Genet. 1995;9:395–400. - PubMed
-
- Chen C, Kim JJ, Thompson RF, Tonegawa S. Hippocampal lesions impair contextual fear conditioning in two strains of mice. Behav Neurosci. 1996;110:1177–1180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials