

Natural genetic exchange between Haemophilus and Neisseria: intergeneric transfer of chromosomal genes between major human pathogens
- PMID: 9770495
- PMCID: PMC22840
- DOI: 10.1073/pnas.95.21.12381
Natural genetic exchange between Haemophilus and Neisseria: intergeneric transfer of chromosomal genes between major human pathogens
Abstract
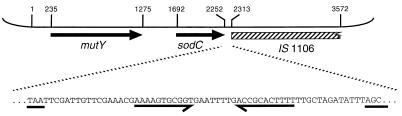
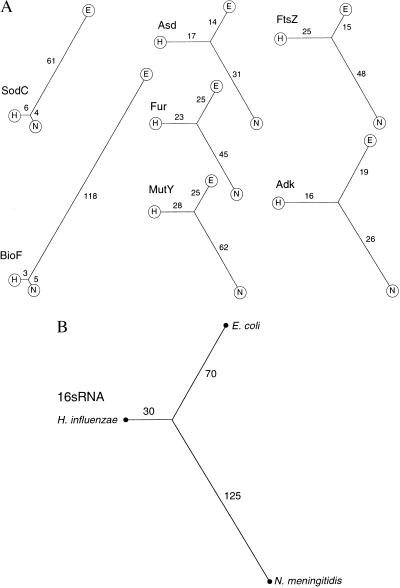
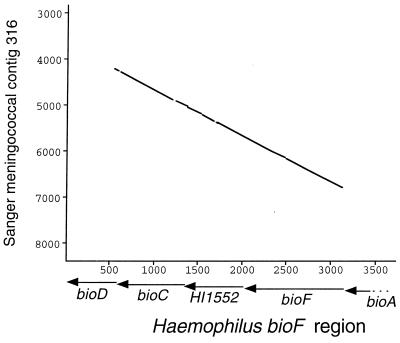
Members of the bacterial families Haemophilus and Neisseria, important human pathogens that commonly colonize the nasopharynx, are naturally competent for DNA uptake from their environment. In each genus this process is discriminant in favor of its own and against foreign DNA through sequence specificity of DNA receptors. The Haemophilus DNA uptake apparatus binds a 29-bp oligonucleotide domain containing a highly conserved 9-bp core sequence, whereas the neisserial apparatus binds a 10-bp motif. Each motif ("uptake sequence", US) is highly over-represented in the chromosome of the corresponding genus, particularly concentrated with core sequences in inverted pairs forming gene terminators. Two Haemophilus core USs were unexpectedly found forming the terminator of sodC in Neisseria meningitidis (meningococcus), and sequence analysis strongly suggests that this virulence gene, located next to IS1106, arose through horizontal transfer from Haemophilus. By using USs as search strings in a computer-based analysis of genome sequence, it was established that while USs of the "wrong" genus do not occur commonly in Neisseria or Haemophilus, where they do they are highly likely to flag domains of chromosomal DNA that have been transferred from Haemophilus. Three independent domains of Haemophilus-like DNA were found in the meningococcal chromosome, associated respectively with the virulence gene sodC, the bio gene cluster, and an unidentified orf. This report identifies intergenerically transferred DNA and its source in bacteria, and further identifies transformation with heterologous chromosomal DNA as a way of establishing potentially important chromosomal mosaicism in these pathogenic bacteria.
Figures


References
-
- Kilian M. J Gen Microbiol. 1976;93:9–62. - PubMed
-
- Danner D B, Deich R A, Sisco K L, Smith H O. Gene. 1980;11:311–318. - PubMed
-
- Smith H O, Tomb J F, Dougherty B A, Fleischmann R D, Venter J C. Science. 1995;269:538–540. - PubMed
-
- Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, Kerlavage A R, Bult C J, Tomb J F, Dougherty B A, Merrick J M, et al. Science. 1995;269:496–512. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
