

Deficient DNA-ligase activity in the metabolic disease tyrosinemia type I
- PMID: 9770534
- PMCID: PMC22879
- DOI: 10.1073/pnas.95.21.12614
Deficient DNA-ligase activity in the metabolic disease tyrosinemia type I
Abstract
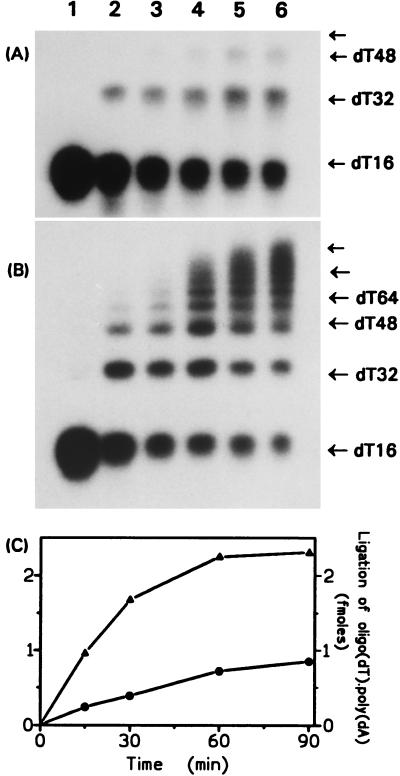
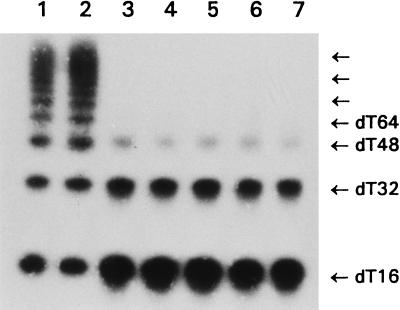
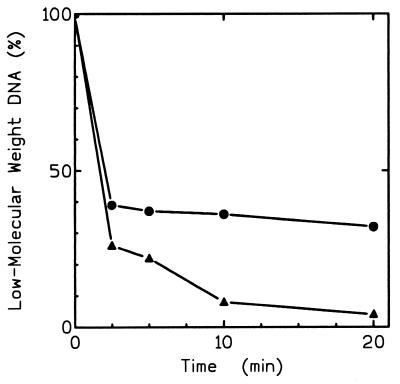
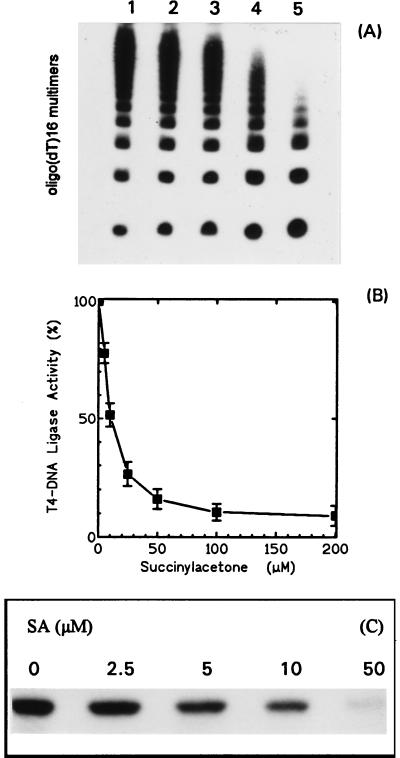
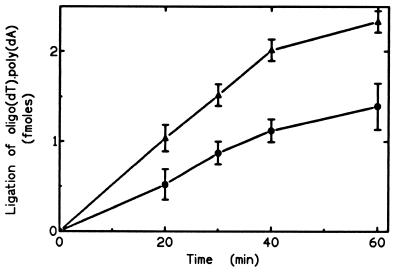
Hereditary tyrosinemia type I (HT1) is an autosomal recessive inborn error of metabolism caused by the deficiency of fumarylacetoacetate hydrolase, the last enzyme in the tyrosine catabolism pathway. This defect results in accumulation of succinylacetone (SA) that reacts with amino acids and proteins to form stable adducts via Schiff base formation, lysine being the most reactive amino acid. HT1 patients surviving beyond infancy are at considerable risk for the development of hepatocellular carcinoma, and a high level of chromosomal breakage is observed in HT1 cells, suggesting a defect in the processing of DNA. In this paper we show that the overall DNA-ligase activity is low in HT1 cells (about 20% of the normal value) and that Okazaki fragments are rejoined at a reduced rate compared with normal fibroblasts. No mutation was found by sequencing the ligase I cDNA from HT1 cells, and the level of expression of the ligase I mRNA was similar in normal and HT1 fibroblasts, suggesting the presence of a ligase inhibitor. SA was shown to inhibit in vitro the overall DNA-ligase activity present in normal cell extracts. The activity of purified T4 DNA-ligase, whose active site is also a lysine residue, was inhibited by SA in a dose-dependent manner. These results suggest that accumulation of SA reduces the overall ligase activity in HT1 cells and indicate that metabolism errors may play a role in regulating enzymatic activities involved in DNA replication and repair.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
