

Not just scaffolding: plectin regulates actin dynamics in cultured cells
- PMID: 9808630
- PMCID: PMC317224
- DOI: 10.1101/gad.12.21.3442
Not just scaffolding: plectin regulates actin dynamics in cultured cells
Abstract
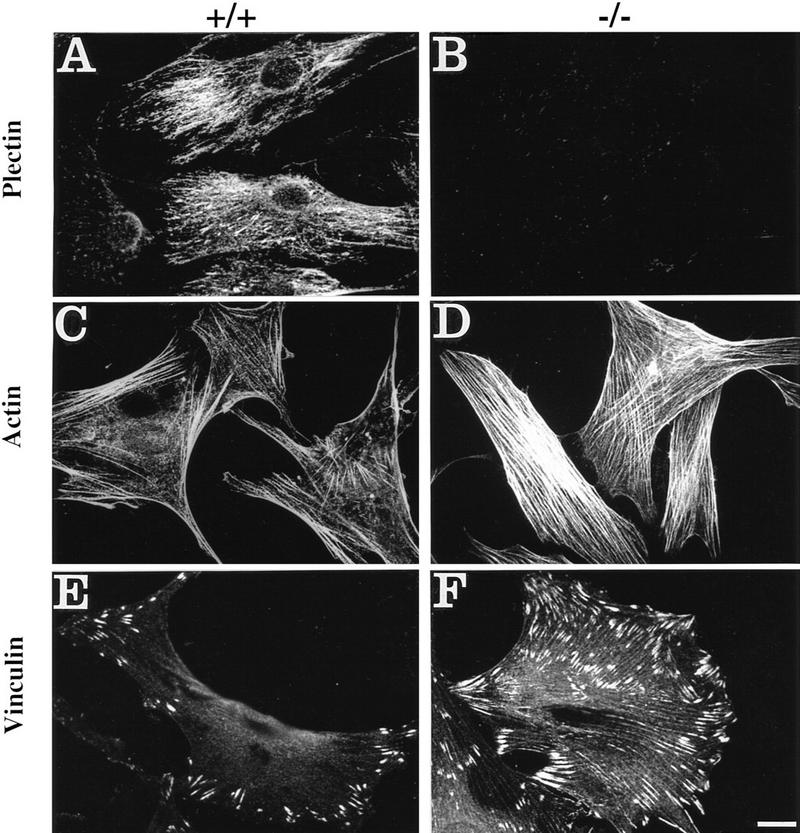
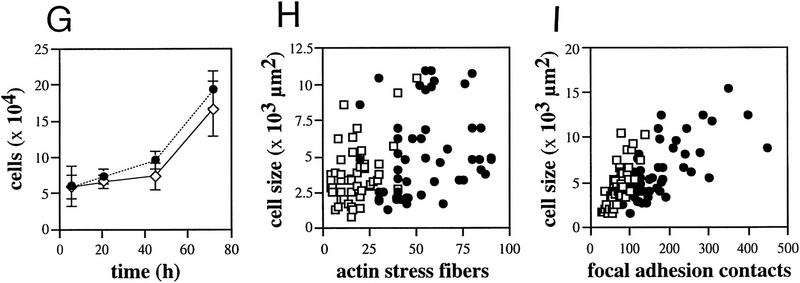
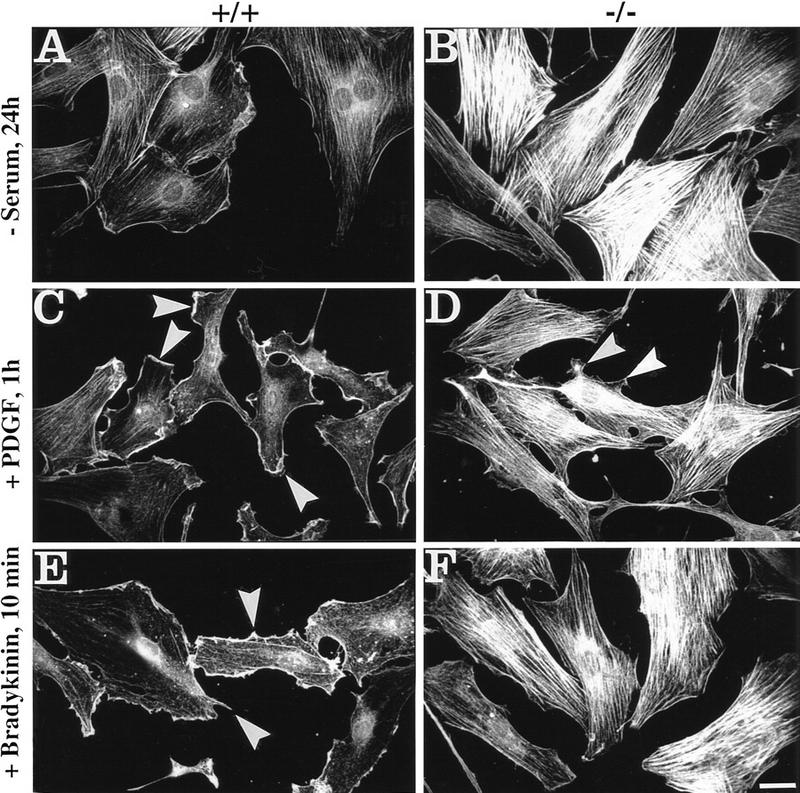
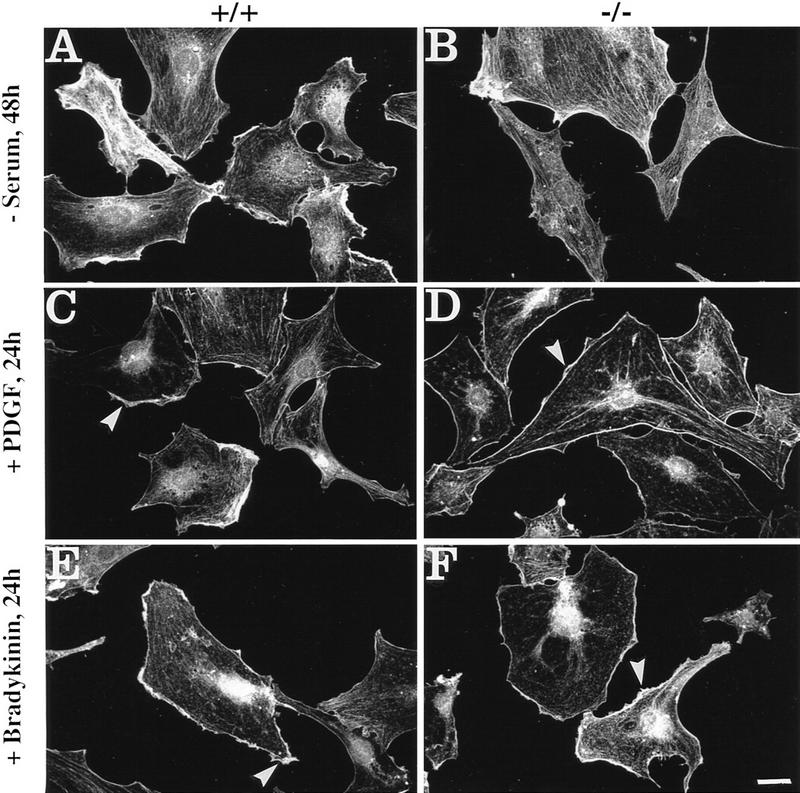
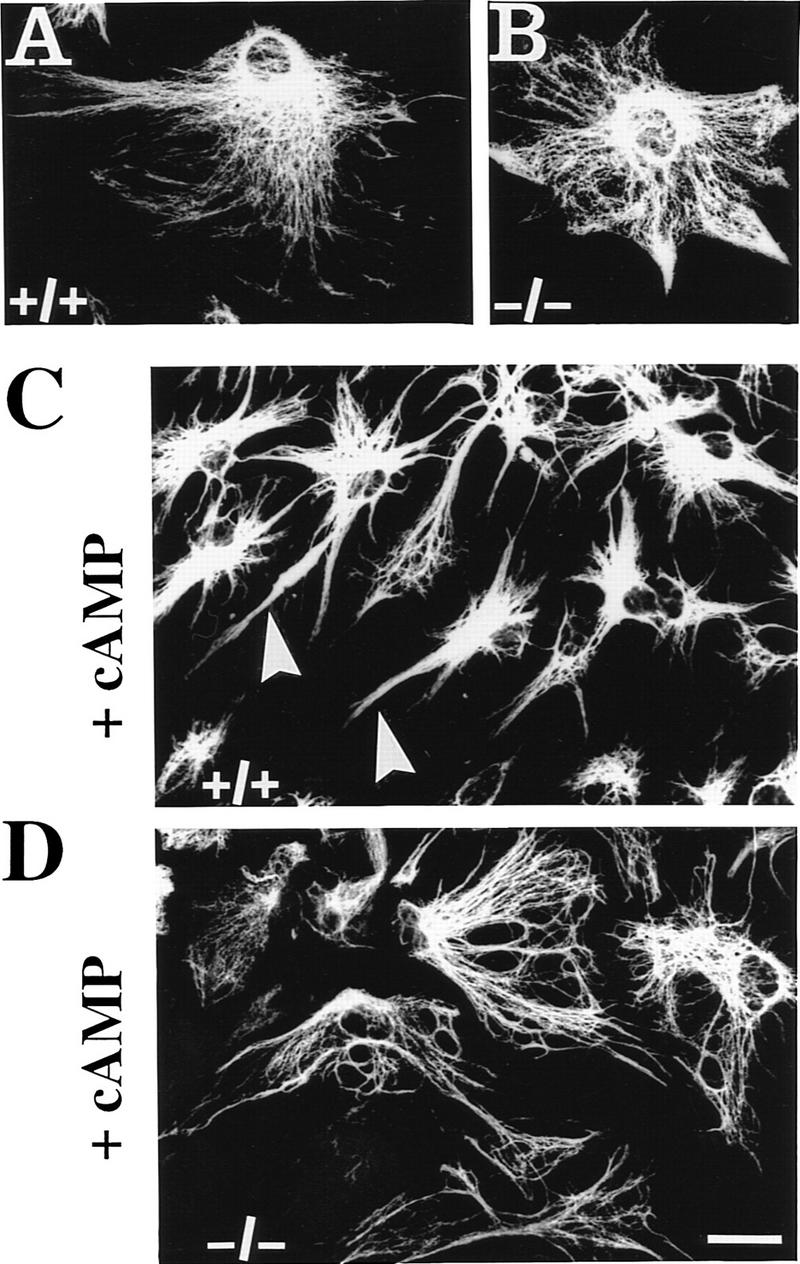
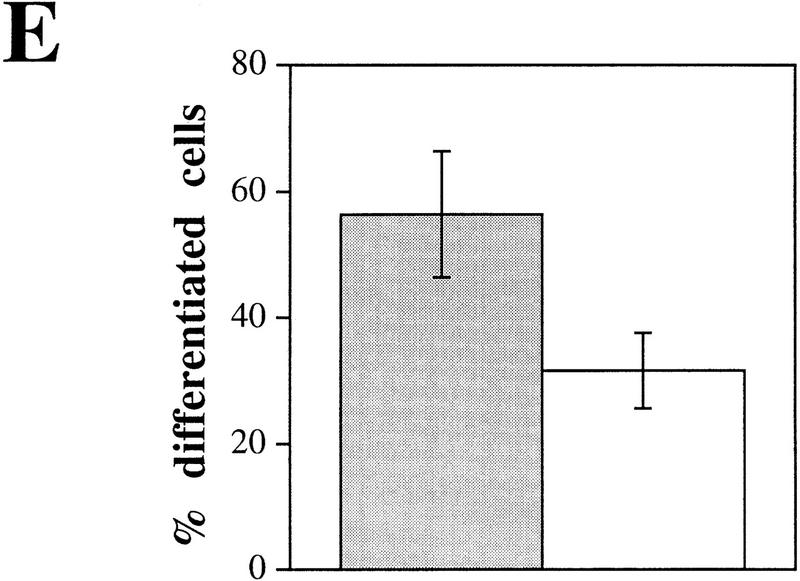
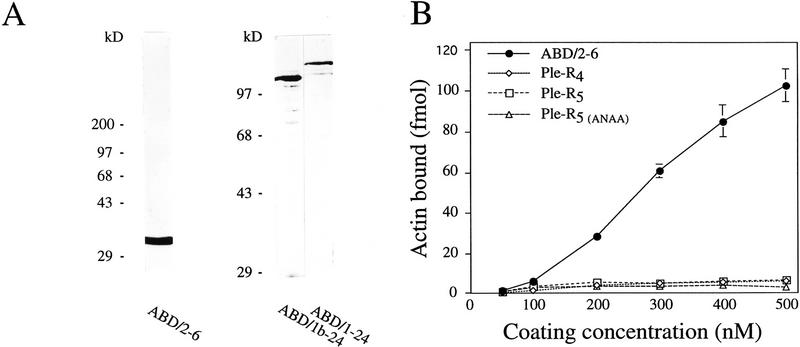
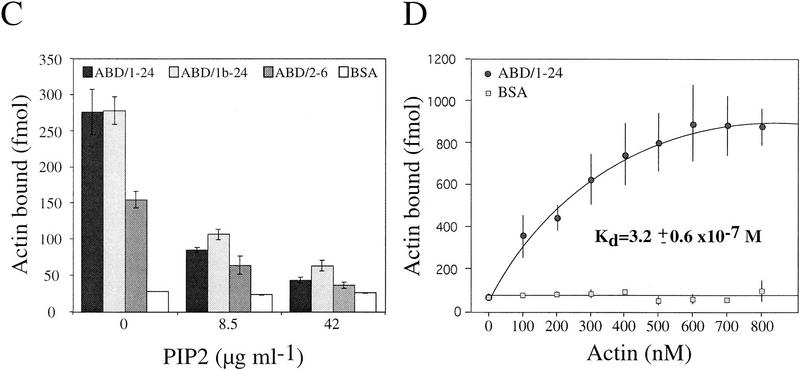
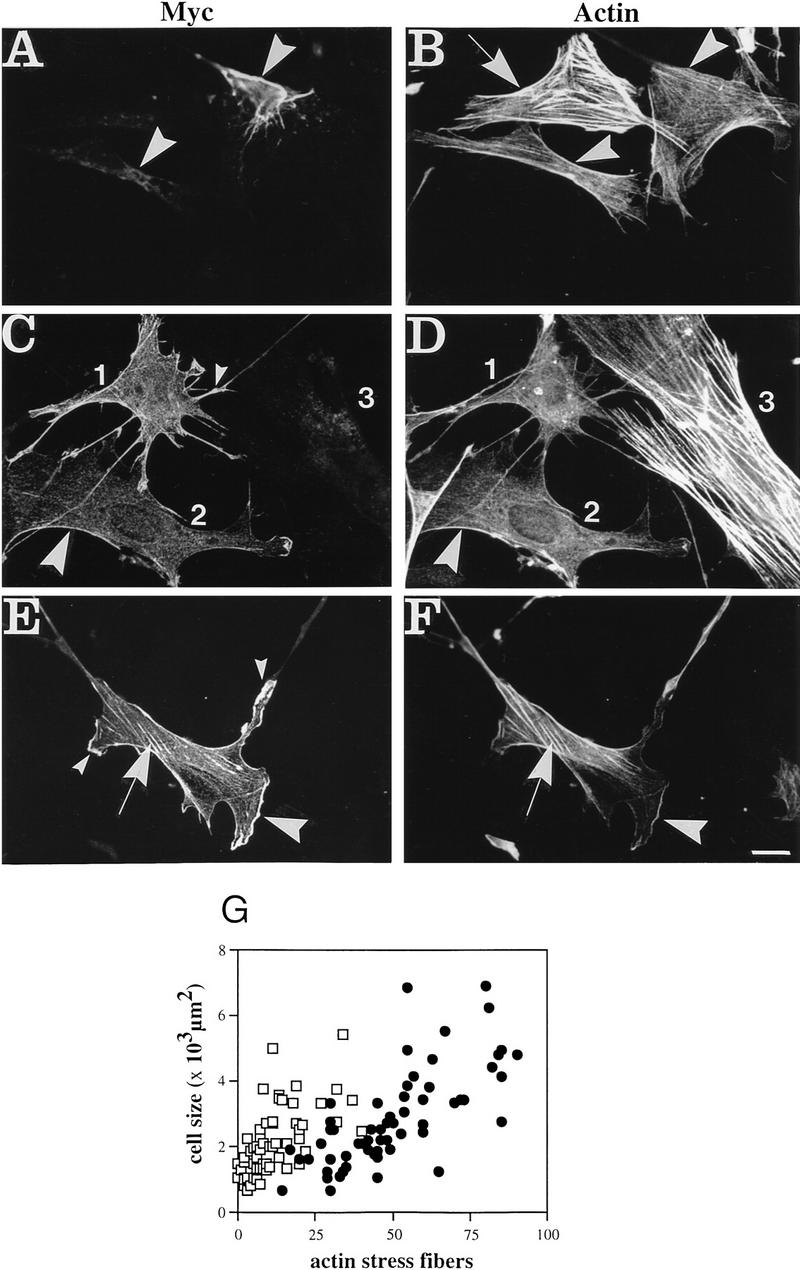
Plectin, a major linker and scaffolding protein of the cytoskeleton, has been shown to be essential for the mechanical integrity of skin, skeletal muscle, and heart. Studying fibroblast and astroglial cell cultures derived from plectin (-/-) mice, we found that their actin cytoskeleton, including focal adhesion contacts, was developed more extensively than in wild-type cells. Also it failed to show characteristic short-term rearrangments in response to extracellular stimuli activating the Rho/Rac/Cdc42 signaling cascades. As a consequence, cell motility, adherence, and shear stress resistance were altered, and morphogenic processes were delayed. Furthermore, we show that plectin interacts with G-actin in vitro in a phosphatidylinositol-4,5-biphosphate-dependent manner and associates with actin stress fibers in living cells. The actin stress fiber phenotype of plectin-deficient fibroblasts could be reversed to a large degree by transient transfection of full-length plectin or plectin fragments containing the amino-terminal actin-binding domain (ABD). These results reveal a novel role of plectin as regulator of cellular processes involving actin filament dynamics that goes beyond its proposed role in scaffolding and mechanical stabilization of cells.
Figures
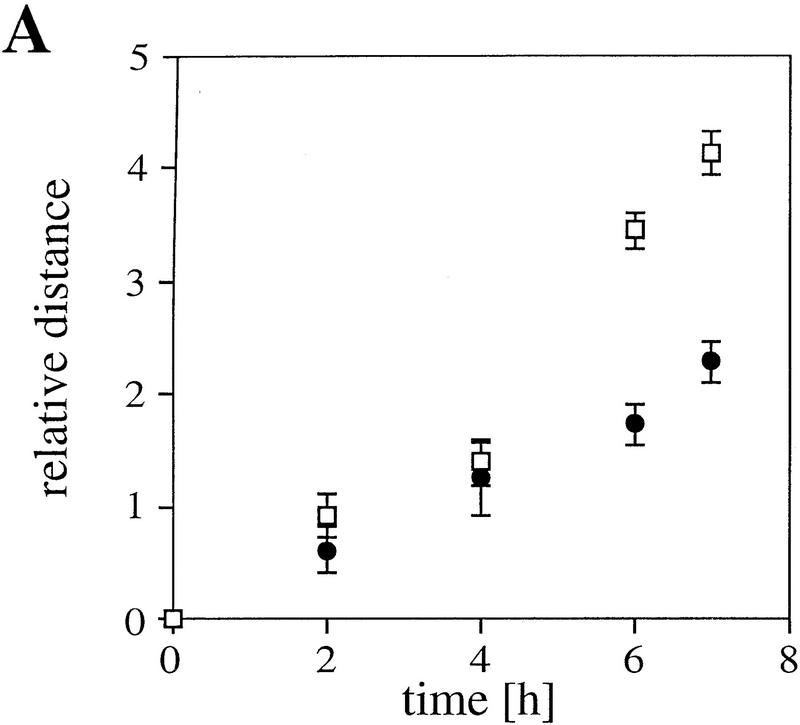
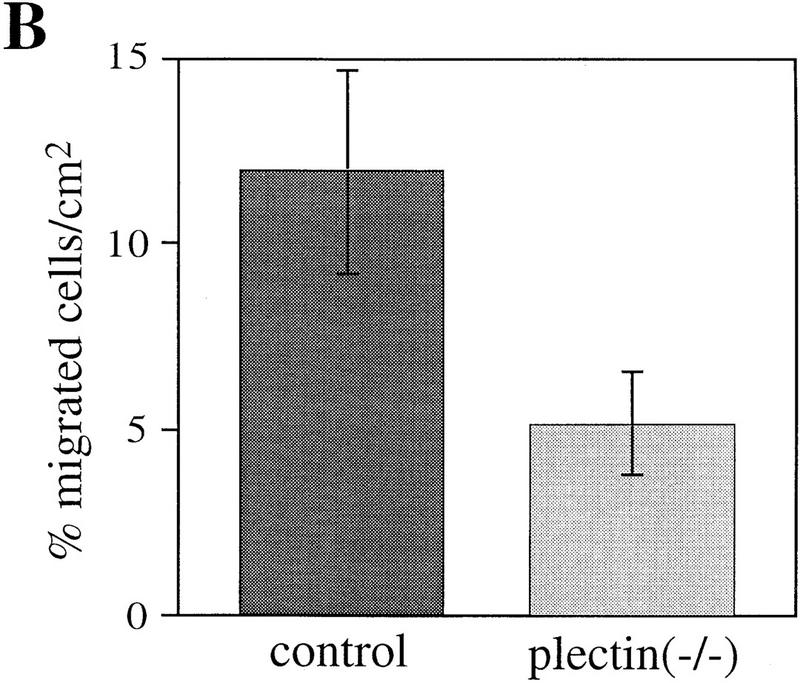
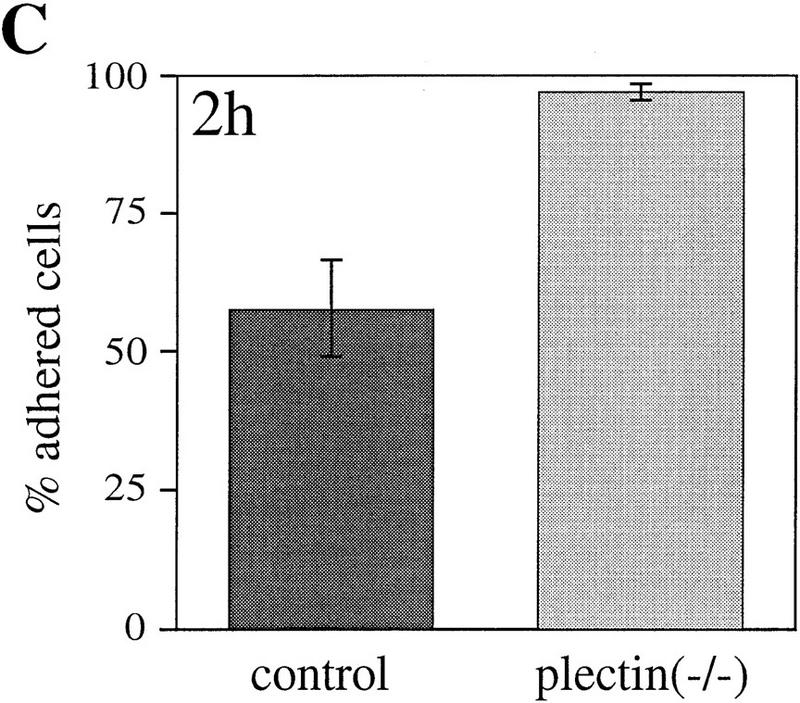
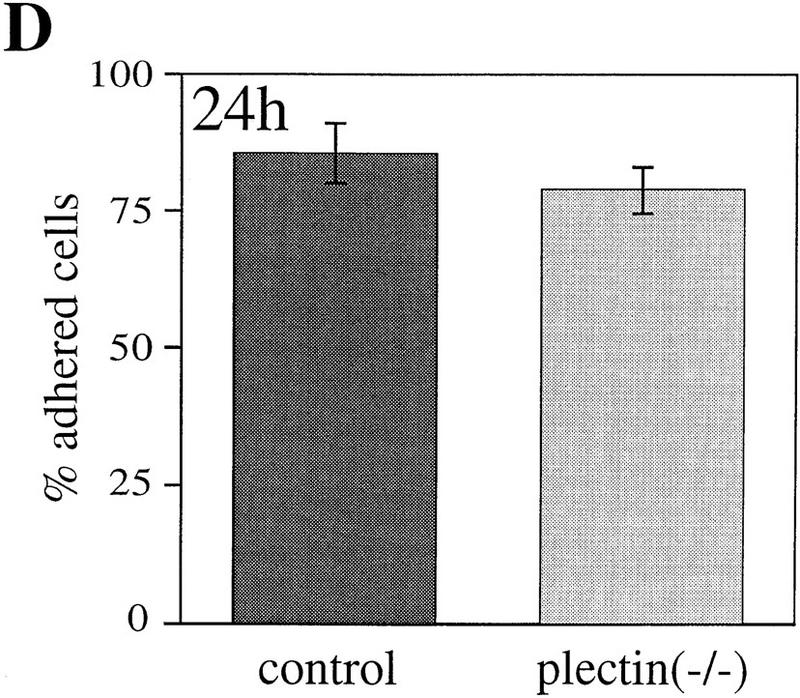
References
-
- Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Involvement of microtubules in the control of adhesion-dependent signal transduction. Curr Biol. 1996;6:1279–1289. - PubMed
-
- Cunningham CC, Stossel TP, Kwiatkowski DJ. Enhanced motility in NIH 3T3 fibroblasts that overexpress gelsolin. Science. 1991;251:1233–1236. - PubMed
-
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous