

Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy
- PMID: 9811342
- PMCID: PMC1853408
- DOI: 10.1016/S0002-9440(10)65738-0
Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy
Abstract
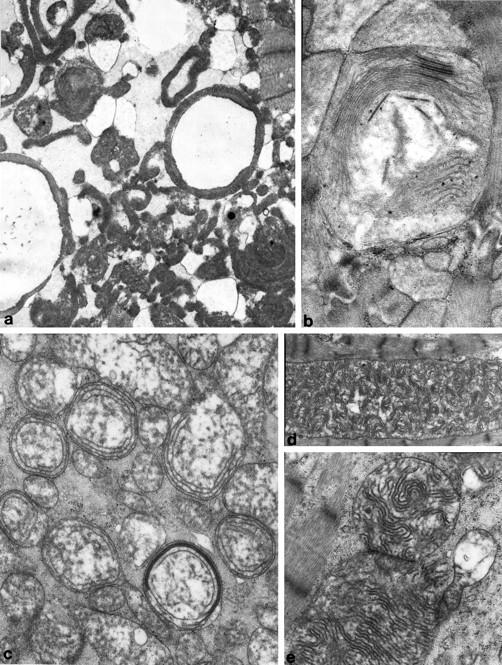
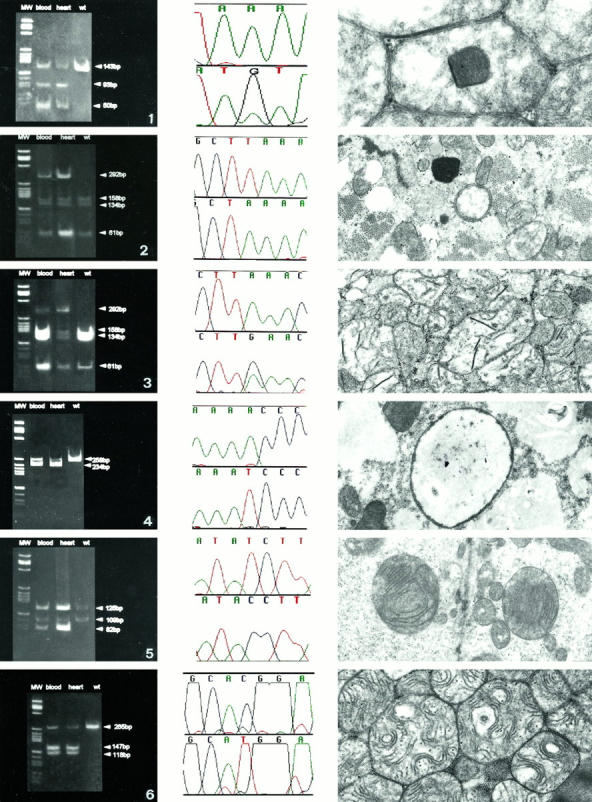
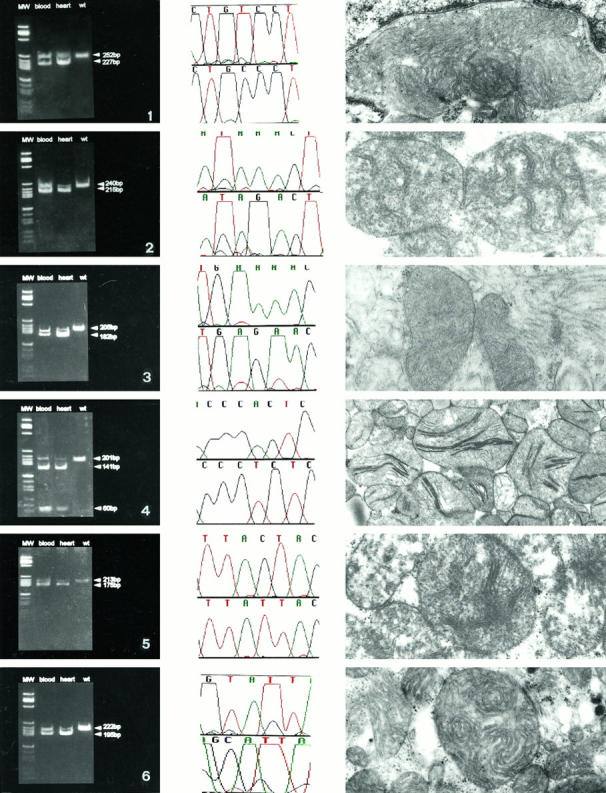
Mitochondrial (mt)DNA defects, both deletions and tRNA point mutations, have been associated with cardiomyopathies. The aim of the study was to determine the prevalence of pathological mtDNA mutations and to assess associated defects of mitochondrial enzyme activity in dilated cardiomyopathy (DCM) patients with ultrastructural abnormalities of cardiac mitochondria. In a large cohort of 601 DCM patients we performed conventional light and electron microscopy on endomyocardial biopsy samples. Cases with giant organelles, angulated, tubular, and concentric cristae, and crystalloid or osmiophilic inclusion bodies were selected for mtDNA analysis. Mutation screening techniques, automated DNA sequencing, restriction enzyme digestion, and densitometric assays were performed to identify mtDNA mutations, assess heteroplasmy, and quantify the amount of mutant in myocardial and blood DNA. Of 601 patients (16 to 63 years; mean, 43.5 +/- 12.7 years), 85 had ultrastructural evidence of giant organelles, with abnormal cristae and inclusion bodies; 19 of 85 (22.35%) had heteroplasmic mtDNA mutations (9 tRNA, 5 rRNA, and 4 missense, one in two patients) that were not found in 111 normal controls and in 32 DCM patients without the above ultrastructural mitochondrial abnormalities. In all cases, the amount of mutant was higher in heart than in blood. In hearts of patients that later underwent transplantation, cytochrome c oxidase (Cox) activity was significantly lower in cases with mutations than in those without or controls (P = 0.0008). NADH dehydrogenase activity was only slightly reduced in cases with mutations (P = 0.0388), whereas succinic dehydrogenase activity did not significantly differ between DCM patients with mtDNA mutations and those without or controls. The present study represents the first attempt to detect a morphological, easily identifiable marker to guide mtDNA mutation screening. Pathological mtDNA mutations are associated with ultrastructurally abnormal mitochondria, and reduced Cox activity in a small subgroup of non-otherwise-defined, idiopathic DCMs, in which mtDNA defects may constitute the basis for, or contribute to, the development of congestive heart failure.
Figures
References
-
- Ozawa T, Tanaka M, Sugiyama S, Hattori K, Ito K, Ohno K, Takahashi A, Sato W, Takada G, Mayumi B: Multiple mitochondrial DNA deletions exist in cardiomyocytes of patients with hypertrophic or dilated cardiomyopathy. Biochem Biophys Res Commun 1990, 170:830-836 - PubMed
-
- Casali C, Santorelli F, D’Amati G, Bernucci P, DeBiase L, DiMauro S: Novel mtDNA point mutation in maternally inherited cardiomyopathy. Biochem Biophys Res Commun 1995, 213:588-593 - PubMed
-
- Merante F, Myint T, Tein I, Benson L, Robinson BH: An additional mitochondrial tRNAIle point mutation (A-to-G at nucleotide 4295) causing hypertrophic cardiomyopathy. Hum Mutat 1996, 8:216-222 - PubMed
-
- Zeviani M, Cellera C, Antozzi C, Rimoldi M, Morandi L, Villani F, Tiranti V, DiDonato S: Maternally inherited myopathy and cardiomyopathy: association with mutation in mitochondrial DNA tRNALeu(UUR). Lancet 1991, 338:143-147 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
