

Large-scale protein structure modeling of the Saccharomyces cerevisiae genome
- PMID: 9811845
- PMCID: PMC24864
- DOI: 10.1073/pnas.95.23.13597
Large-scale protein structure modeling of the Saccharomyces cerevisiae genome
Abstract
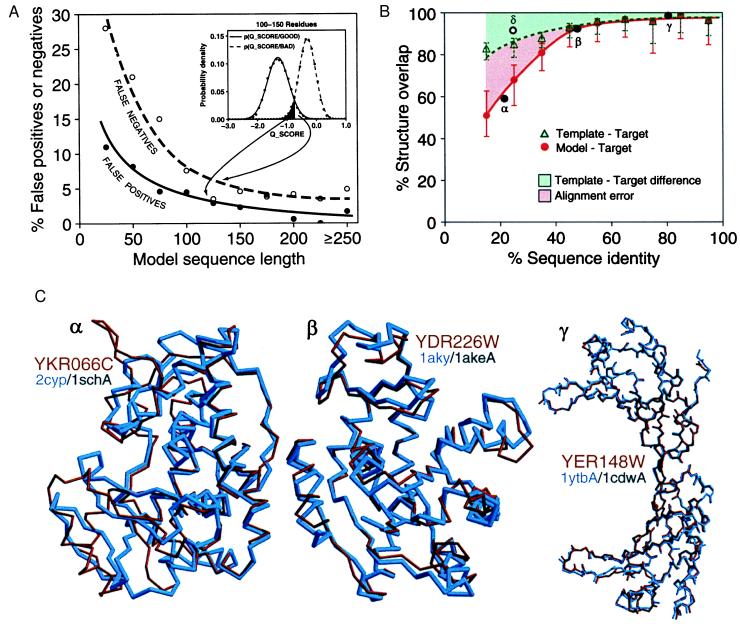
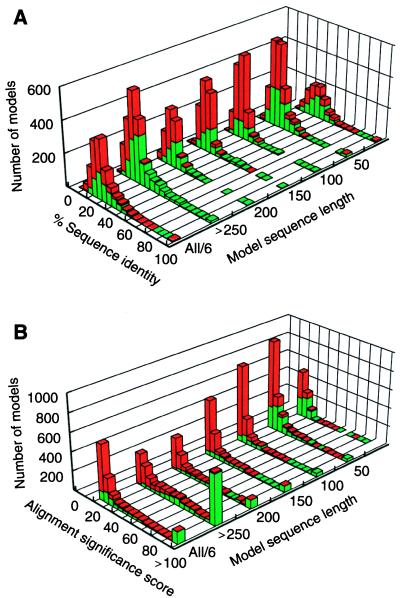
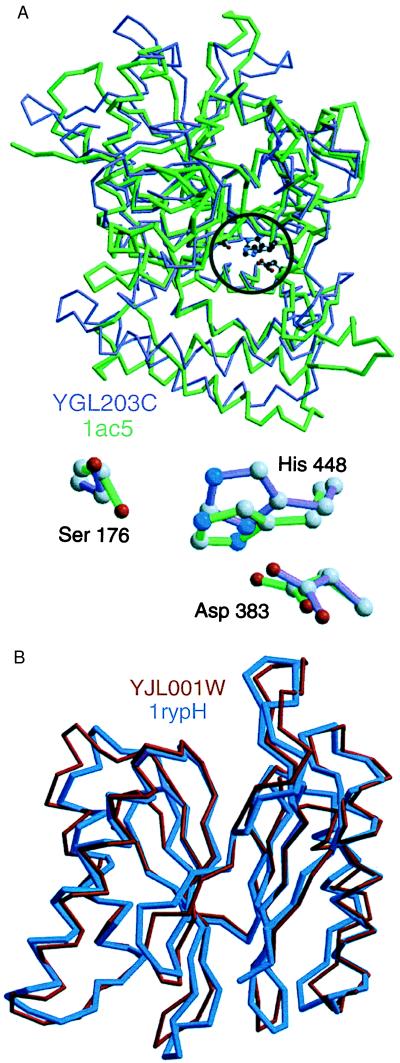
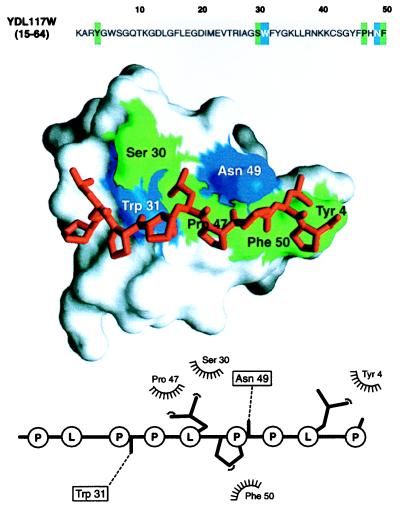
The function of a protein generally is determined by its three-dimensional (3D) structure. Thus, it would be useful to know the 3D structure of the thousands of protein sequences that are emerging from the many genome projects. To this end, fold assignment, comparative protein structure modeling, and model evaluation were automated completely. As an illustration, the method was applied to the proteins in the Saccharomyces cerevisiae (baker's yeast) genome. It resulted in all-atom 3D models for substantial segments of 1,071 (17%) of the yeast proteins, only 40 of which have had their 3D structure determined experimentally. Of the 1,071 modeled yeast proteins, 236 were related clearly to a protein of known structure for the first time; 41 of these previously have not been characterized at all.
Figures
References
-
- Oliver S G. Nature (London) 1996;379:597–600. - PubMed
-
- Koonin E V, Mushegian A R. Curr Opin Gen Dev. 1996;6:757–762. - PubMed
-
- Dujon B. Trends Genet. 1996;12:263–270. - PubMed
-
- Orengo C A, Jones D T, Thornton J M. Nature (London) 1994;372:631–634. - PubMed
-
- Miklos G L G, Rubin G M. Cell. 1996;86:521–529. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
