

Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting
- PMID: 9811863
- PMCID: PMC24882
- DOI: 10.1073/pnas.95.23.13698
Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting
Abstract
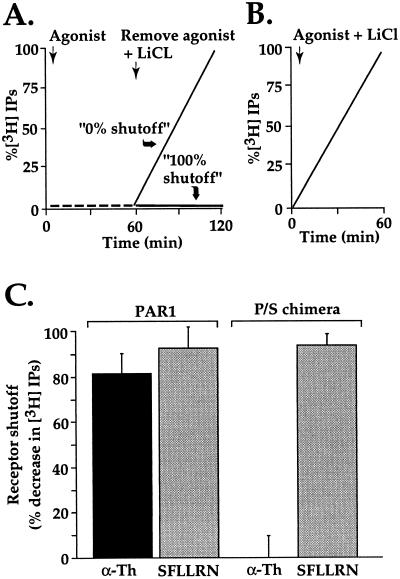
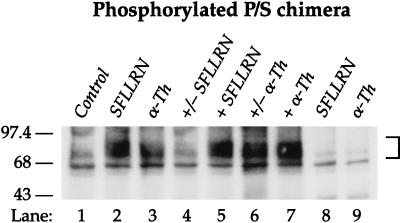
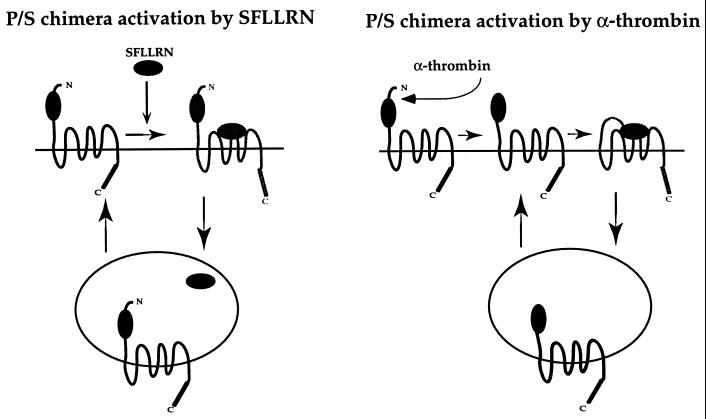
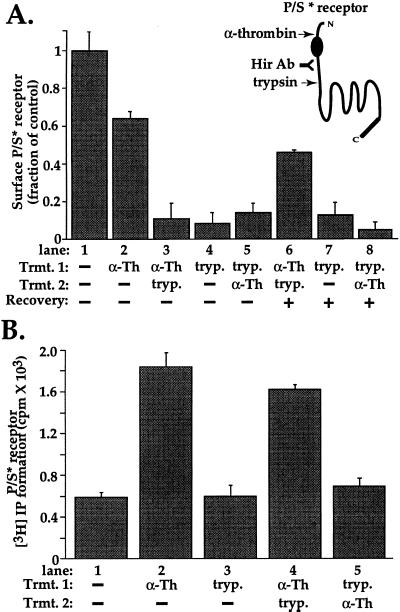
The irreversible proteolytic mechanism by which protease-activated receptor-1 (PAR1), the G protein-coupled receptor (GPCR) for thrombin, is activated raises the question of how it is shut off. Like classic GPCRs, activated PAR1 is rapidly phosphorylated and internalized, but unlike classic GPCRs, which recycle, internalized PAR1 is sorted to lysosomes. A chimeric PAR1 bearing the substance P receptor's cytoplasmic carboxyl tail sequestered and recycled like wild-type substance P receptor. In cells expressing this chimera, signaling in response to the PAR1-activating peptide SFLLRN ceased as expected upon removal of this agonist. Strikingly, however, when the chimera was activated proteolytically by thrombin, signaling persisted even after thrombin was removed. This persistent signaling was apparently due to "resignaling" by previously activated receptors that had internalized and recycled back to the cell surface. Thus the cytoplasmic carboxyl tail of PAR1 specifies an intracellular sorting pattern that is linked to its signaling properties. In striking contrast to most GPCRs, sorting of activated PAR1 to lysosomes rather than recycling is critical for terminating PAR1 signaling-a trafficking solution to a signaling problem.
Figures
References
-
- Yu S S, Lefkowitz R J, Hausdorff W P. J Biol Chem. 1993;268:337–341. - PubMed
-
- Krueger K M, Daaka Y, Pitcher J A, Lefkowitz R J. J Biol Chem. 1997;272:5–8. - PubMed
-
- Pippig S, Andexinger S, Lohse M J. Mol Pharmacol. 1995;47:666–676. - PubMed
-
- Freedman N J, Lefkowitz R J. Recent Prog Horm Res. 1996;51:319–351. ; discussion 352–353. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
