

Uncoupling signal transducers from oncogenic MET mutants abrogates cell transformation and inhibits invasive growth
- PMID: 9826708
- PMCID: PMC24381
- DOI: 10.1073/pnas.95.24.14379
Uncoupling signal transducers from oncogenic MET mutants abrogates cell transformation and inhibits invasive growth
Abstract
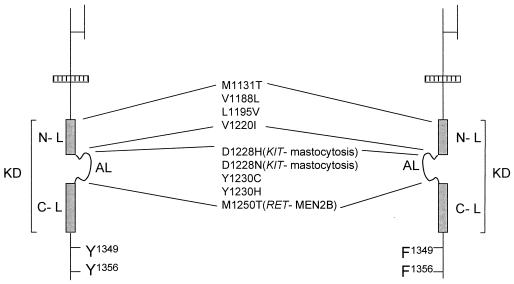
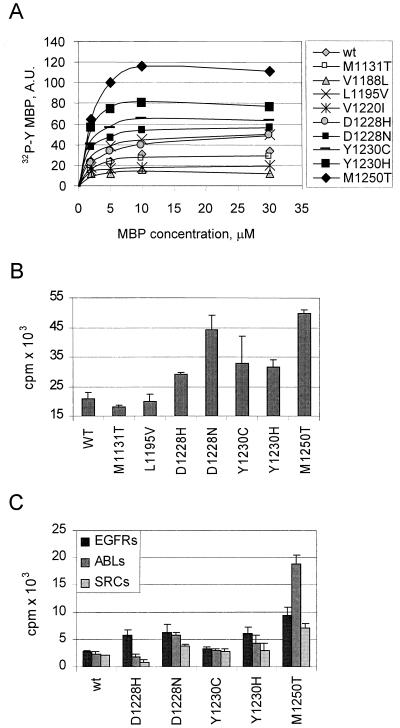
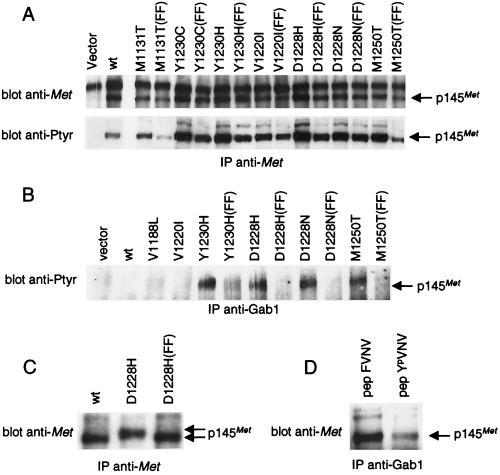
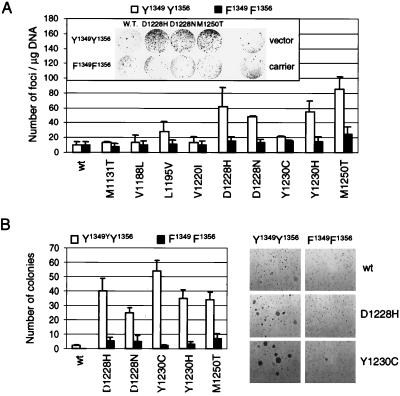
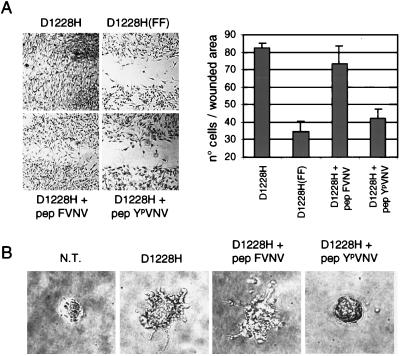
The assumption that genes encoding tyrosine kinase receptors could play a role in human cancers has been confirmed by the identification of oncogenic mutations in the kinase domain of RET and KIT. Recently, homologous residues were found mutated in MET, in papillary renal carcinomas (PRCs). The link coupling these genetic lesions to cellular transformation is still unclear. METPRC mutations result in increased kinase activity and-in some instances, i.e., M1250T substitution-in changes in substrate specificity. A direct correlation occurs between the transforming potential of METPRC mutants and their ability to constitutively associate with signal transducers through two phosphorylated tyrosines (Y1349VHVNATY1356VNV) located in the receptor tail. Substitution of these "docking tyrosines" with phenylalanines leaves unaffected the altered properties of the kinase but abrogates transformation and invasiveness in vitro. Uncoupling the receptor from signal transducers with a tyrosine-phosphorylated peptide derivative (YpVNV) inhibits invasive growth induced by METPRC mutants. These data indicate that constitutive receptor coupling to downstream signal transducers is a key mechanism in neoplastic transformation driven by mutated MET and suggest a therapeutic strategy to target neoplastic diseases associated with this oncogene.
Figures
References
-
- Mulligan L M, Kwok J B, Healey C S, Elsdon M J, Eng C, Gardner E, Love D R, Mole S E, Moore J K, Papi L, et al. Nature (London) 1993;363:458–460. - PubMed
-
- Hofstra R M, Landsvater R M, Ceccherini I, Stulp R P, Stelwagen T, Luo Y, Pasini B, Hoppener J W, van Amstel H K, Romeo G, et al. Nature (London) 1994;367:375–376. - PubMed
-
- Longley B J, Tyrrell L, Lu S Z, Ma Y S, Langley K, Ding T G, Duffy T, Jacobs P, Tang L H, Modlin I. Nat Genet. 1996;12:312–314. - PubMed
-
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, et al. Science. 1998;279:577–580. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
