

The nitric oxide hypothesis of late preconditioning
- PMID: 9833145
- PMCID: PMC3701309
- DOI: 10.1007/s003950050101
The nitric oxide hypothesis of late preconditioning
Abstract
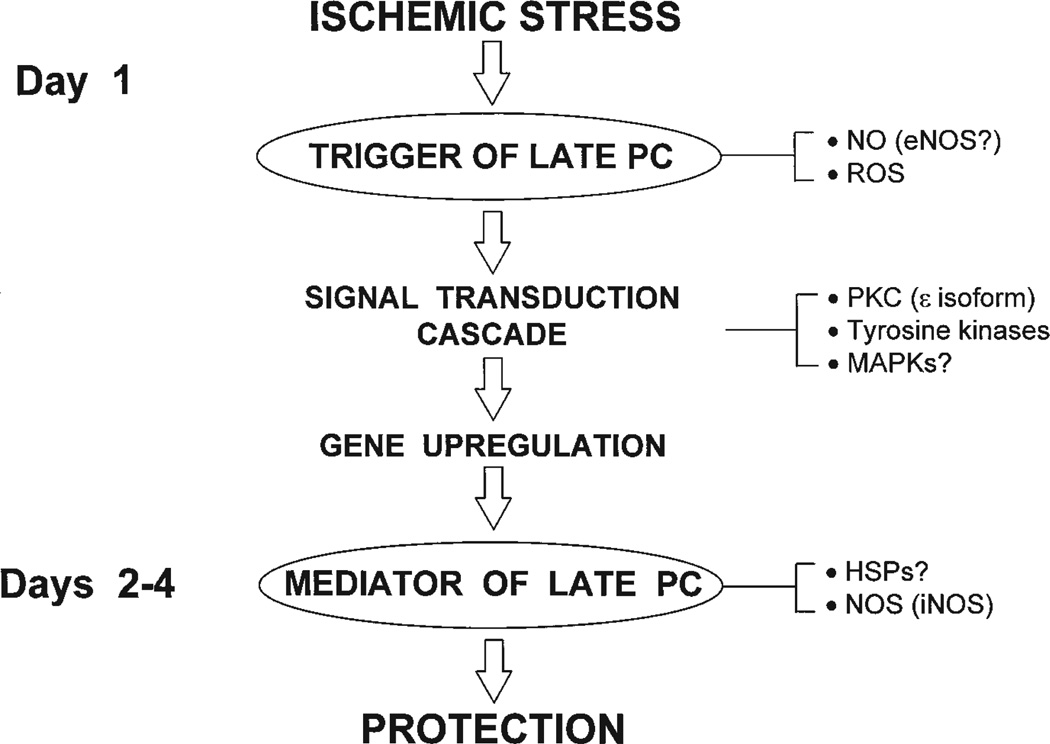
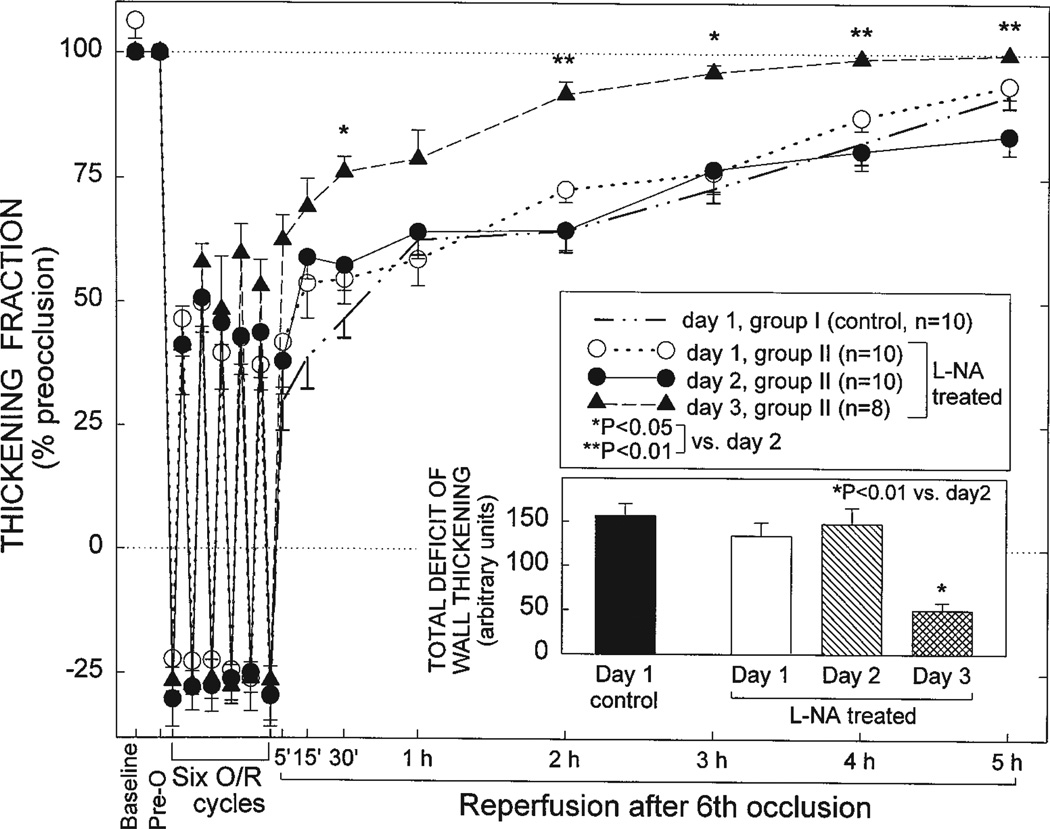
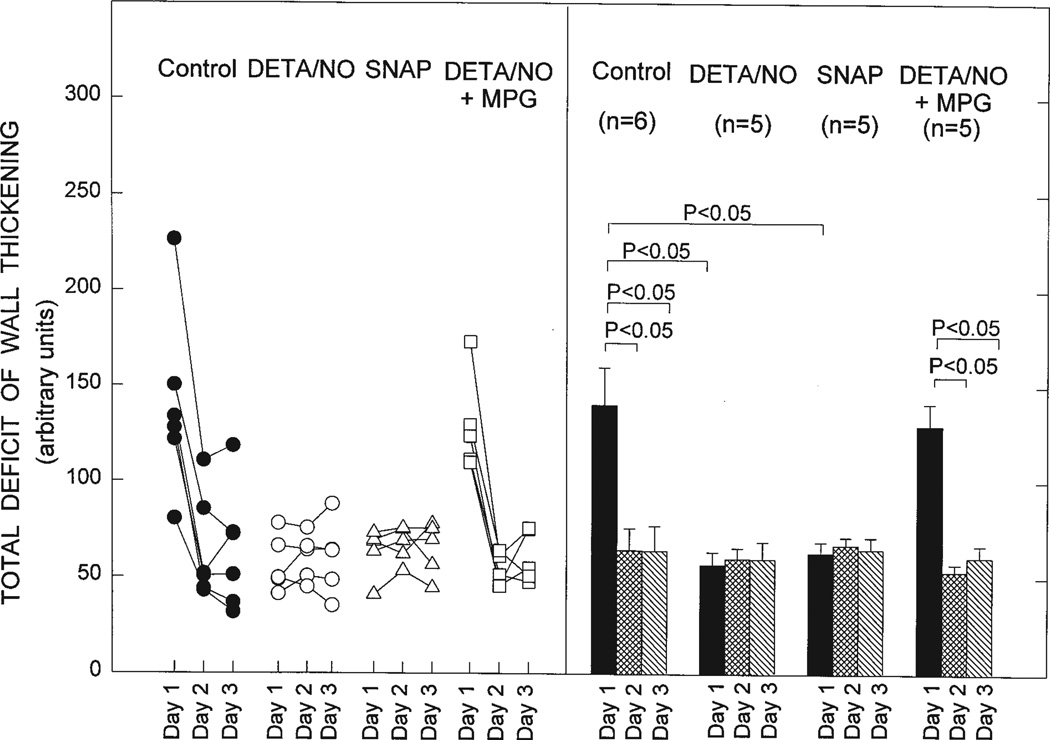
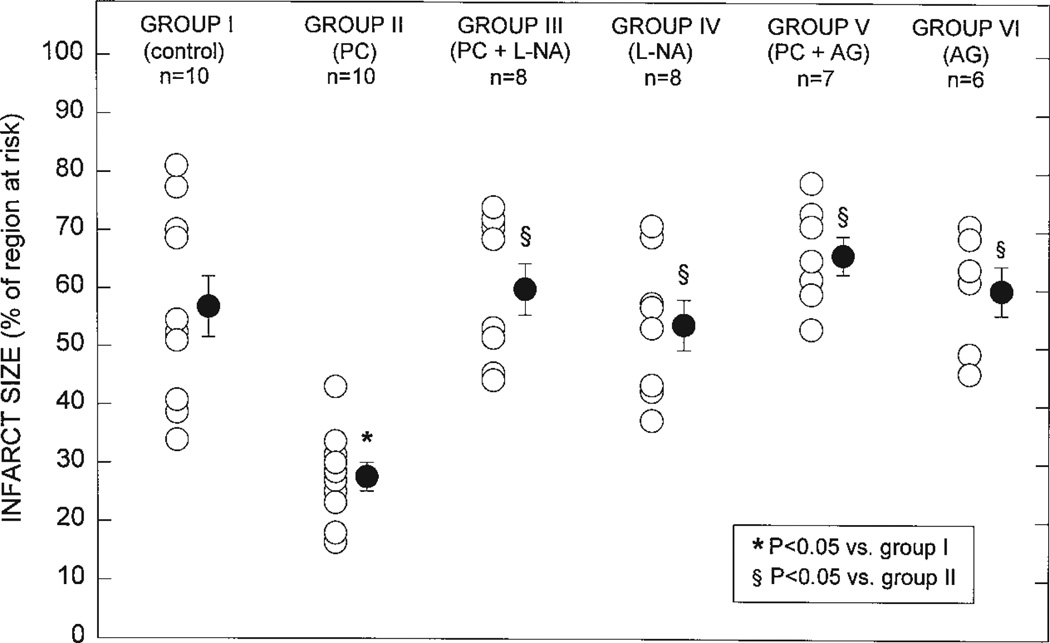
Ischemic preconditioning (PC) occurs in two phases: an early phase, which lasts 2-3 h, and a late phase, which begins 12-24 h later and lasts 3-4 days. The mechanism for the late phase of PC has been the focus of intense investigation. We have recently proposed the "NO hypothesis of late PC", which postulates that NO plays a prominent role both in initiating and in mediating this cardioprotective response. The purpose of this essay is to review the evidence supporting the NO hypothesis of late PC and to discuss its implications. We propose that, on day 1, a brief ischemic stress causes increased production of NO (probably via eNOS) and .O2-, which then react to form ONOO-, ONOO-, in turn, activates the epsilon isoform of protein kinase C (PKC), either directly or via its reactive byproducts such as .OH. Both NO and secondary species derived from .O2- could also stimulate PKC epsilon independently. PKC epsilon activation triggers a complex signaling cascade that involves tyrosine kinases (among which Src and Lck appear to be involved) and probably other kinases, the transcription factor NF-kappa B, and most likely other as yet unknown components, resulting in increased transcription of the iNOS gene and increased iNOS activity on day 2, which is responsible for the protection during the second ischemic challenge. Tyrosine kinases also appear to be involved on day 2, possibly by modulating iNOS activity. According to this paradigm, NO plays two completely different roles in late PC: on day 1, it initiates the development of this response, whereas on day 2, it protects against myocardial ischemia. We propose that two different NOS isoforms are sequentially involved in late PC, with eNOS generating the NO that initiates the development of the PC response on day 1 and iNOS then generating the NO that protects against recurrent ischemia on day 2. The NO hypothesis of late PC puts forth a comprehensive paradigm that can explain both the initiation and the mediation of this complex phenomenon. Besides its pathophysiological implications, this hypothesis has potential clinical reverberations, since NO donors (i.e., nitrates) are widely used clinically and could be used to protect the ischemic myocardium in patients.
Figures





Comment in
-
Pursuit of the role of free radicals in stunning.Basic Res Cardiol. 1998 Oct;93(5):422. Basic Res Cardiol. 1998. PMID: 9935390 No abstract available.
References
-
- Baeuerle PA, Henkel T. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994;12:141–179. - PubMed
-
- Banerjee S, Tang X-L, Qiu Y, Takano H, Manchikalapudi S, Dawn B, Shirk G, Bolli R. Nitroglycerin induces late preconditioning against myocardial stunning via protein kinase C mediated pathway in conscious rabbits. Circulation. 1998 (abstract, in press)
-
- Baxter GF, Marber MS, Patel VC, Yellon DM. Adenosine receptor involvement in a delayed phase of myocardial protection 24 hours after ischemic preconditioning. Circulation. 1994;90:2993–3000. - PubMed
-
- Baxter GF, Goma FM, Yellon DM. Characterisation of the infarct-limiting effect of delayed preconditioning: Time-course and dose-dependency studies in rabbit myocardium. Basic Res Cardiol. 1997;92:159–167. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
