

Recycling of golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering
- PMID: 9852147
- PMCID: PMC2132995
- DOI: 10.1083/jcb.143.6.1505
Recycling of golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering
Abstract
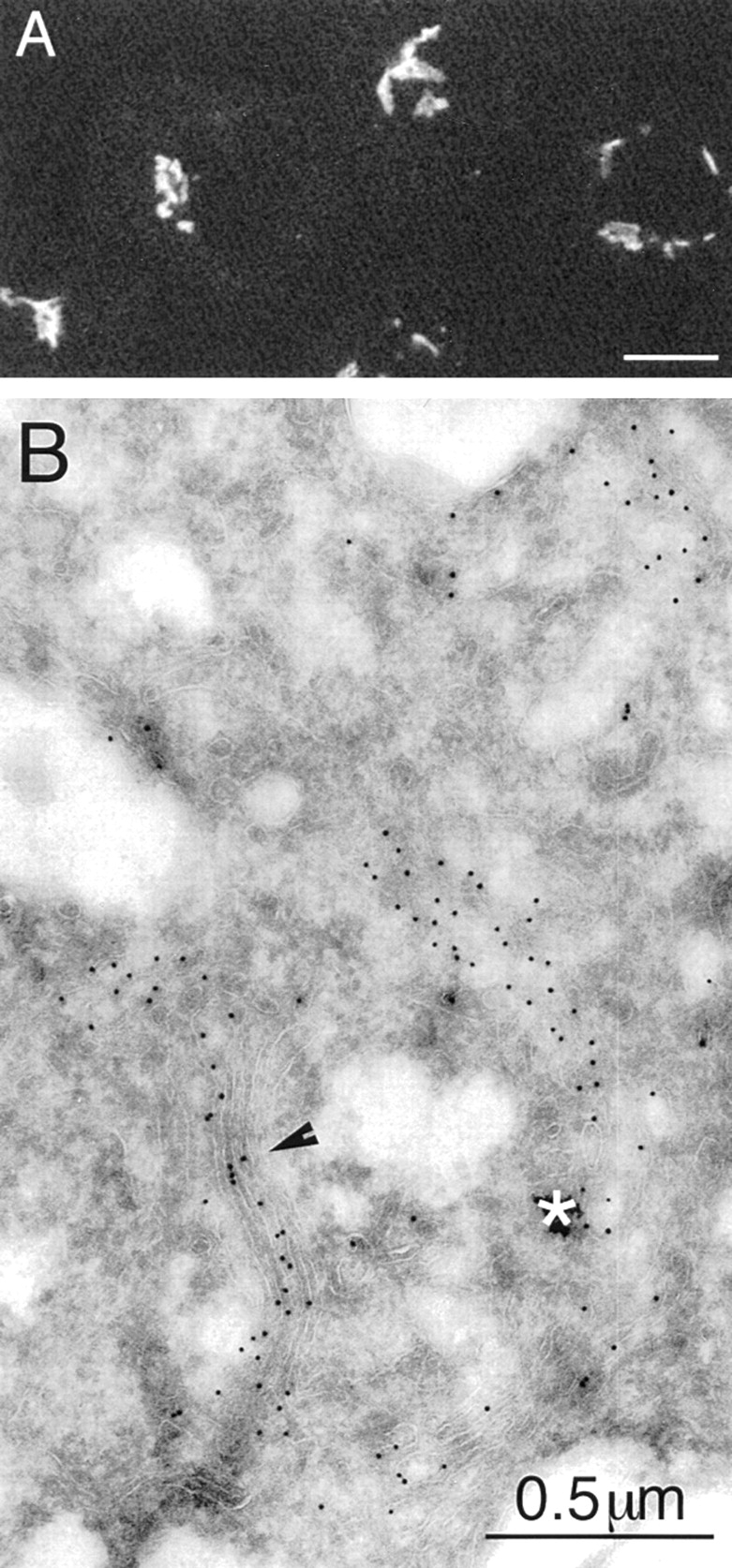
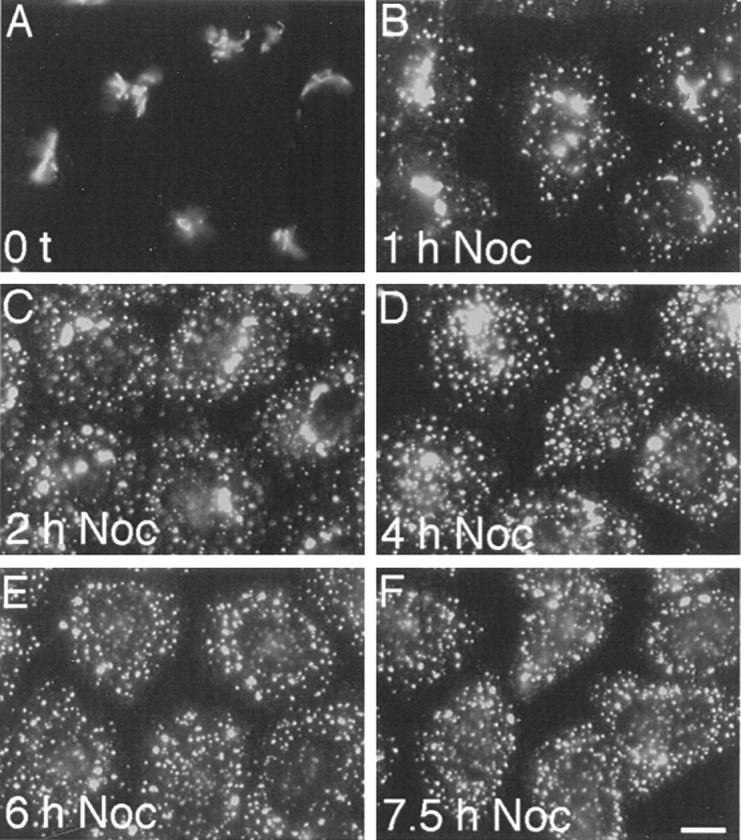
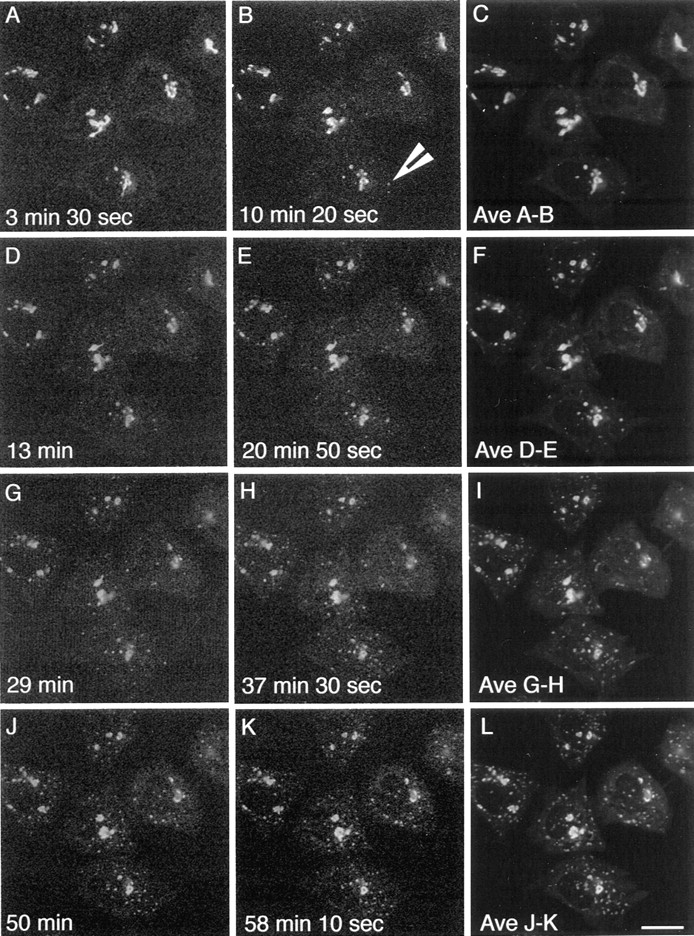
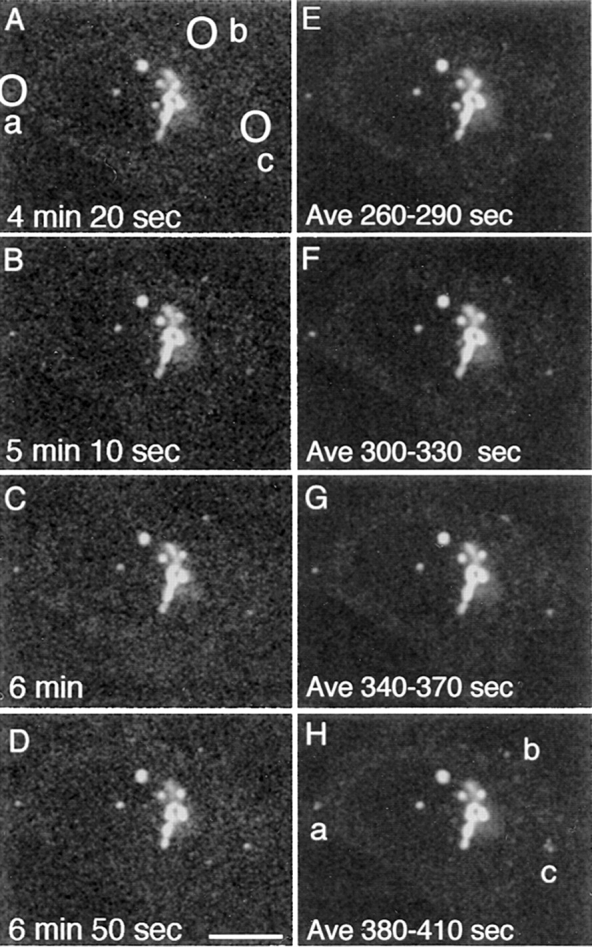
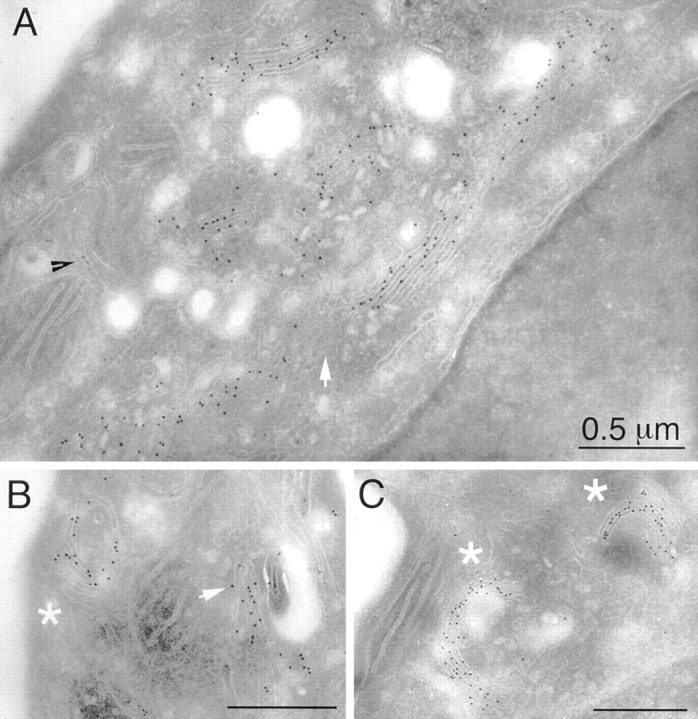
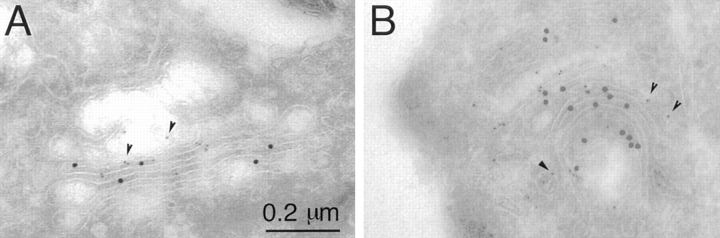
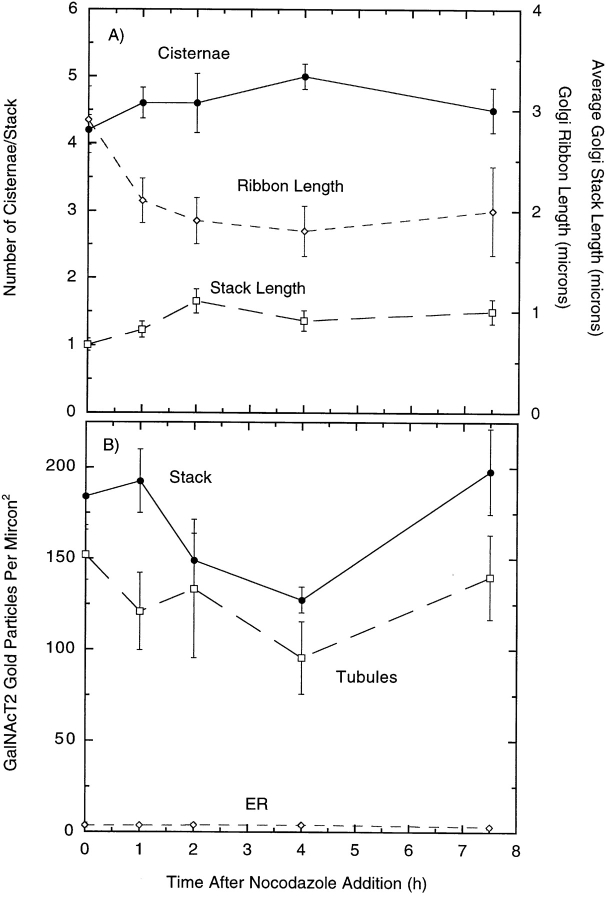
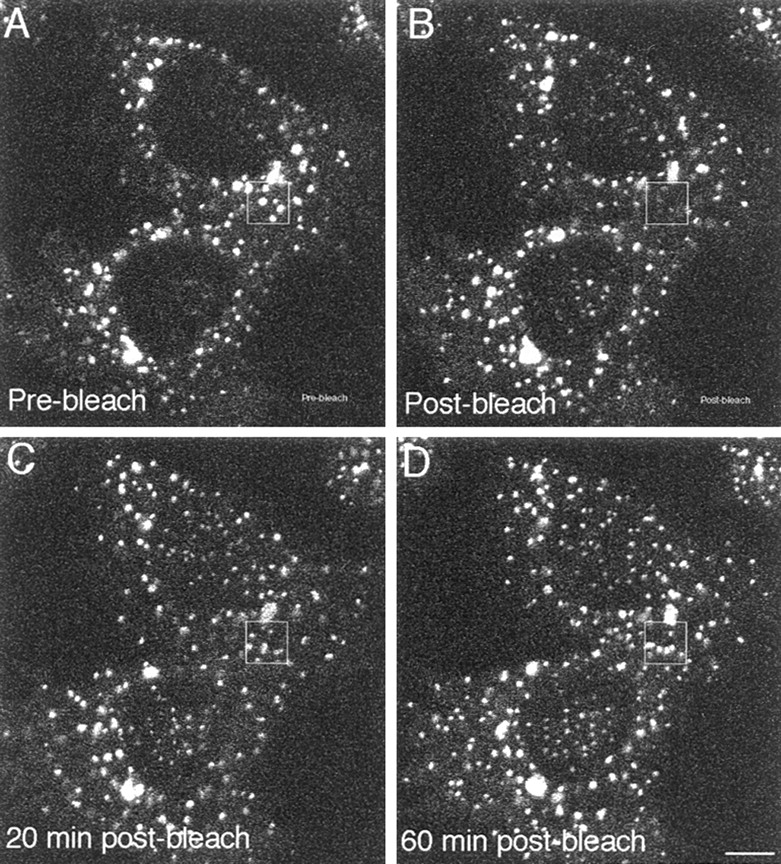
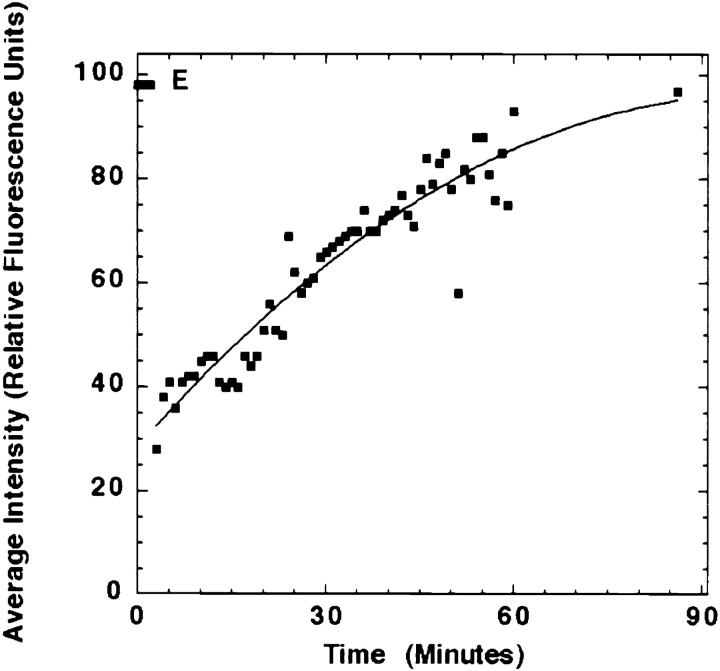
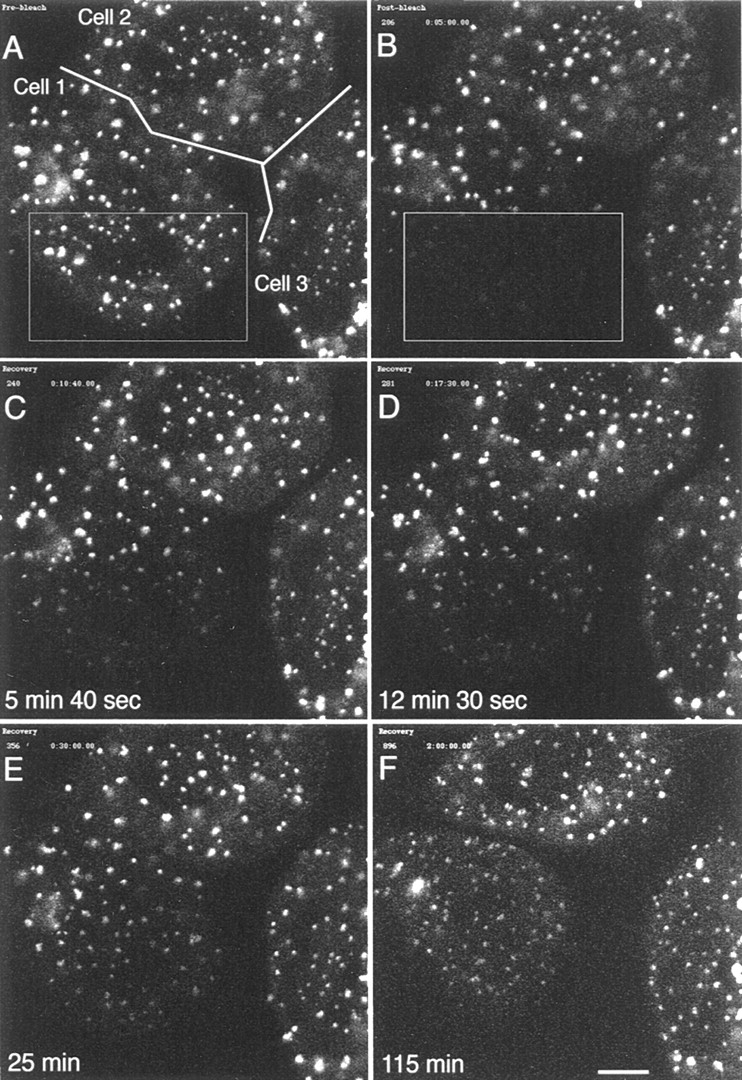
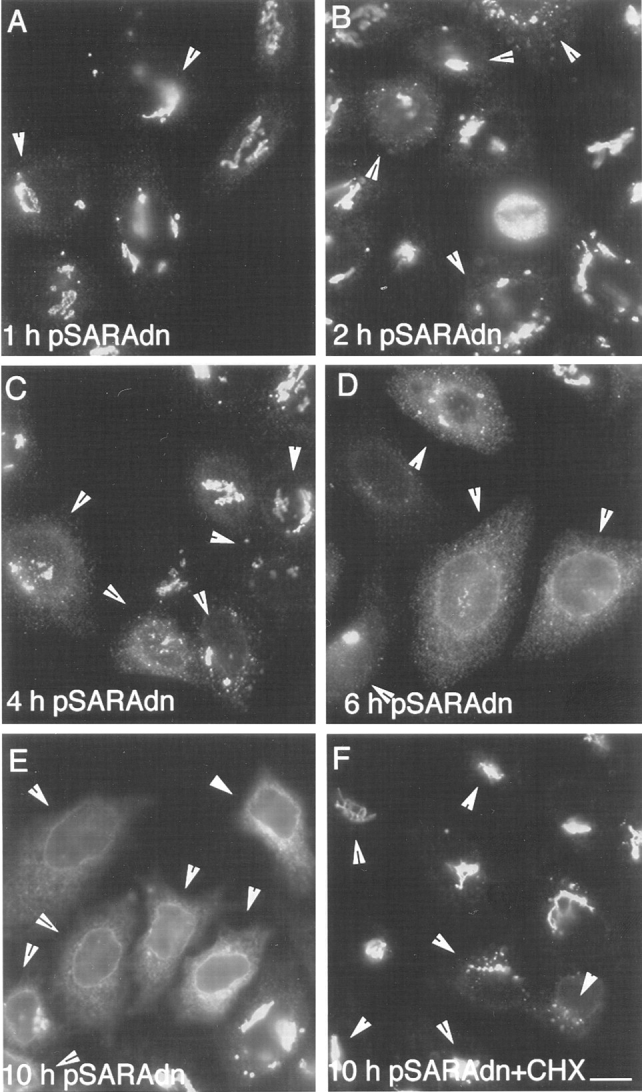
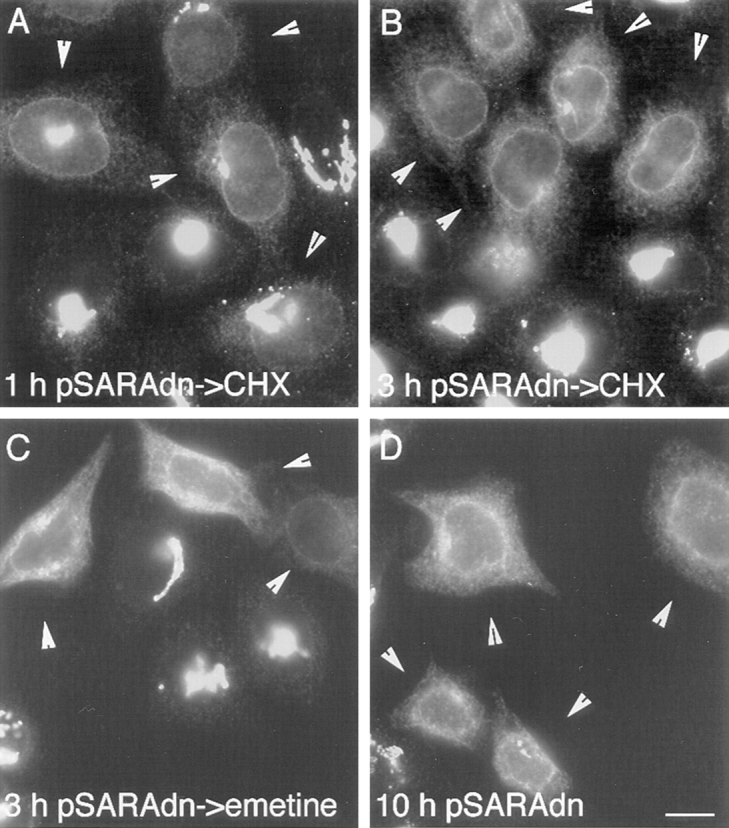
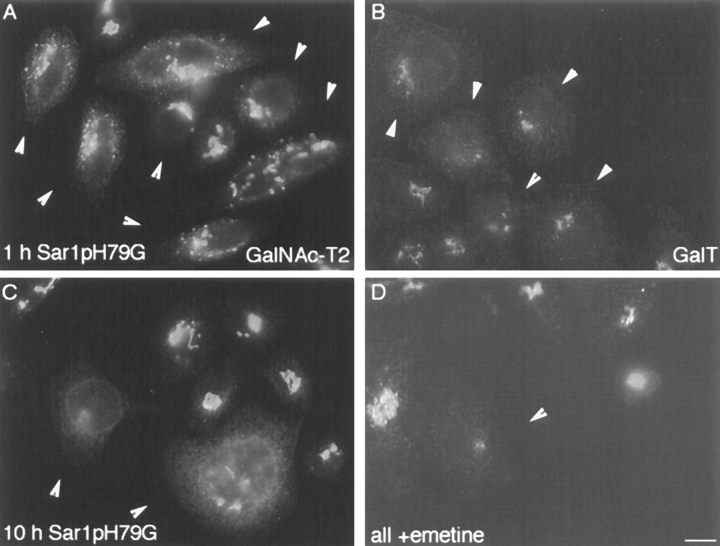
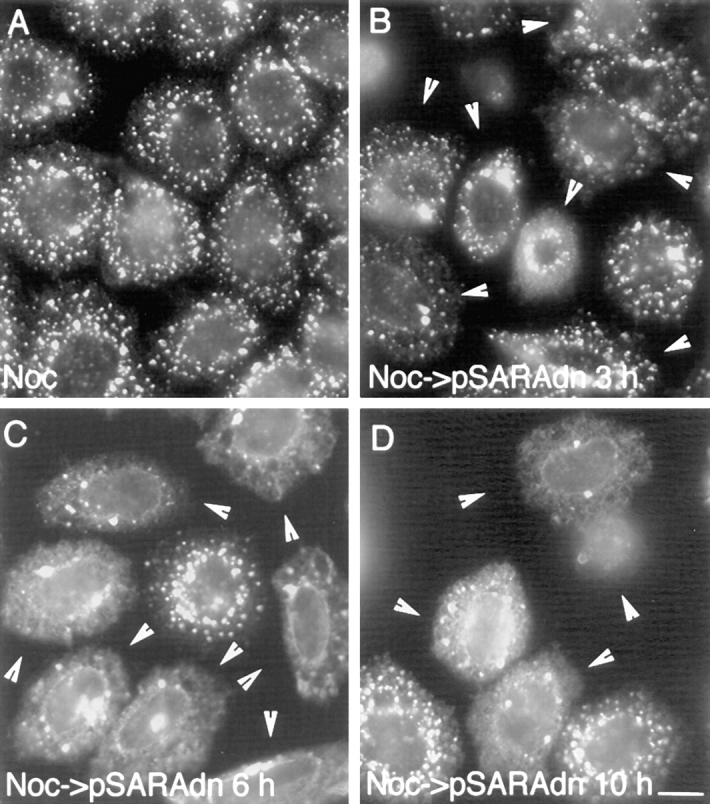
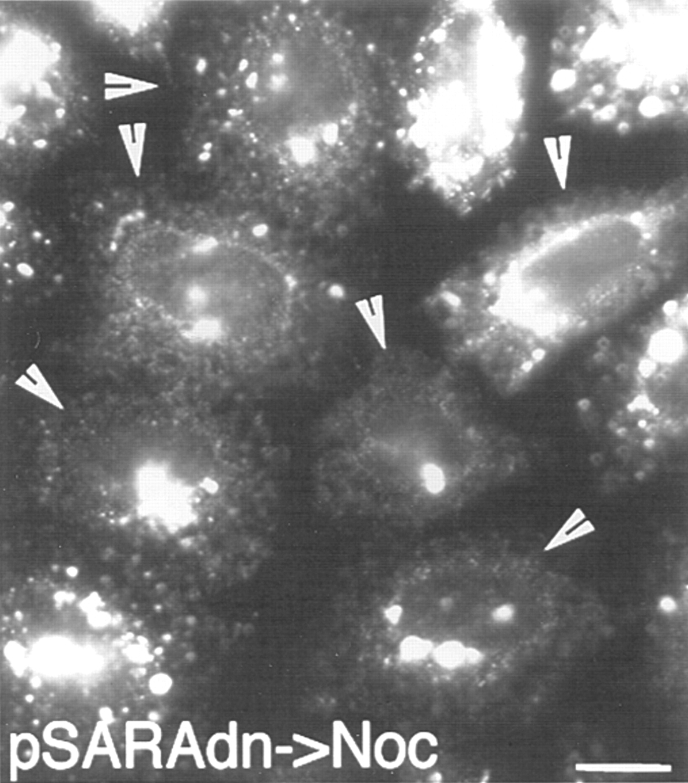
During microtubule depolymerization, the central, juxtanuclear Golgi apparatus scatters to multiple peripheral sites. We have tested here whether such scattering is due to a fragmentation process and subsequent outward tracking of Golgi units or if peripheral Golgi elements reform through a novel recycling pathway. To mark the Golgi in HeLa cells, we stably expressed the Golgi stack enzyme N-acetylgalactosaminyltransferase-2 (GalNAc-T2) fused to the green fluorescent protein (GFP) or to an 11-amino acid epitope, VSV-G (VSV), and the trans/TGN enzyme beta1,4-galactosyltransferase (GalT) fused to GFP. After nocodazole addition, time-lapse microscopy of GalNAc-T2-GFP and GalT-GFP revealed that scattered Golgi elements appeared abruptly and that no Golgi fragments tracked outward from the compact, juxtanuclear Golgi complex. Once formed, the scattered structures were relatively stable in fluorescence intensity for tens of minutes. During the entire process of dispersal, immunogold labeling for GalNAc-T2-VSV and GalT showed that these were continuously concentrated over stacked Golgi cisternae and tubulovesicular Golgi structures similar to untreated cells, suggesting that polarized Golgi stacks reform rapidly at scattered sites. In fluorescence recovery after photobleaching over a narrow (FRAP) or wide area (FRAP-W) experiments, peripheral Golgi stacks continuously exchanged resident proteins with each other through what appeared to be an ER intermediate. That Golgi enzymes cycle through the ER was confirmed by microinjecting the dominant-negative mutant of Sar1 (Sar1pdn) blocking ER export. Sar1pdn was either microinjected into untreated or nocodazole-treated cells in the presence of protein synthesis inhibitors. In both cases, this caused a gradual accumulation of GalNAc-T2-VSV in the ER. Few to no peripheral Golgi elements were seen in the nocodazole-treated cells microinjected with Sar1pdn. In conclusion, we have shown that Golgi-resident glycosylation enzymes recycle through the ER and that this novel pathway is the likely explanation for the nocodazole-induced Golgi scattering observed in interphase cells.
Figures
References
-
- Burkhardt JK. The role of microtubule-based motor proteins in maintaining the structure and function of the Golgi complex. Biochim Biophys Acta. 1998;1404:113–126. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
