

Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation
- PMID: 9861014
- PMCID: PMC28088
- DOI: 10.1073/pnas.95.26.15592
Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation
Abstract
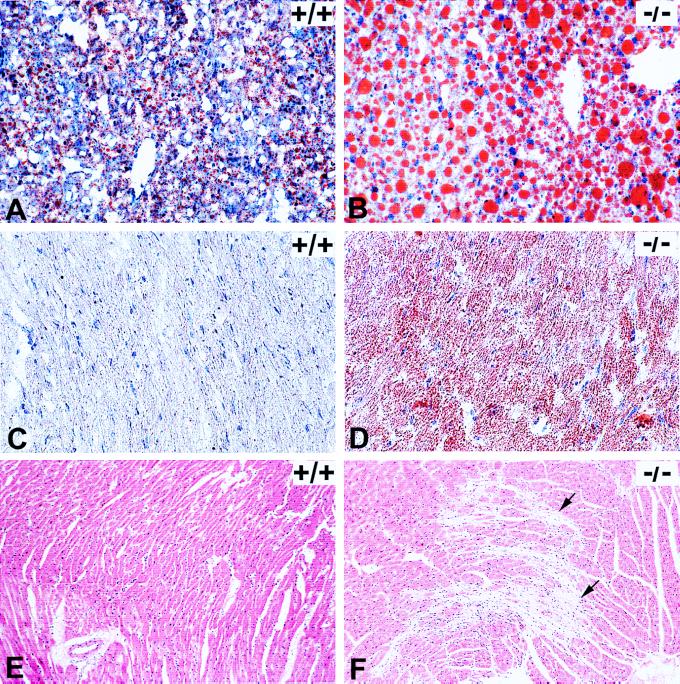
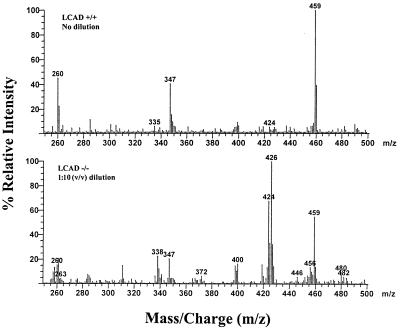
Abnormalities of fatty acid metabolism are recognized to play a significant role in human disease, but the mechanisms remain poorly understood. Long-chain acyl-CoA dehydrogenase (LCAD) catalyzes the initial step in mitochondrial fatty acid oxidation (FAO). We produced a mouse model of LCAD deficiency with severely impaired FAO. Matings between LCAD +/- mice yielded an abnormally low number of LCAD +/- and -/- offspring, indicating frequent gestational loss. LCAD -/- mice that reached birth appeared normal, but had severely reduced fasting tolerance with hepatic and cardiac lipidosis, hypoglycemia, elevated serum free fatty acids, and nonketotic dicarboxylic aciduria. Approximately 10% of adult LCAD -/- males developed cardiomyopathy, and sudden death was observed in 4 of 75 LCAD -/- mice. These results demonstrate the crucial roles of mitochondrial FAO and LCAD in vivo.
Figures

References
-
- Mannaerts G P, Debeer L J. Ann NY Acad Sci. 1982;386:30–39. - PubMed
-
- Nicholls D G, Locke R M. Physiol Rev. 1984;64:1–64. - PubMed
-
- Roe C R, Coates P M. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1995. pp. 1501–1533.
-
- Boles R G, Buck E A, Blitzer M G, Platt M S, Cowan T M, Martin S K, Yoon H, Madsen J A, Reyes-Mugica M, Rinaldo P. J Pediatr (Berlin) 1998;132:924–933. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
