

Severe cardiomyopathy in mice lacking dystrophin and MyoD
- PMID: 9874799
- PMCID: PMC15120
- DOI: 10.1073/pnas.96.1.220
Severe cardiomyopathy in mice lacking dystrophin and MyoD
Abstract
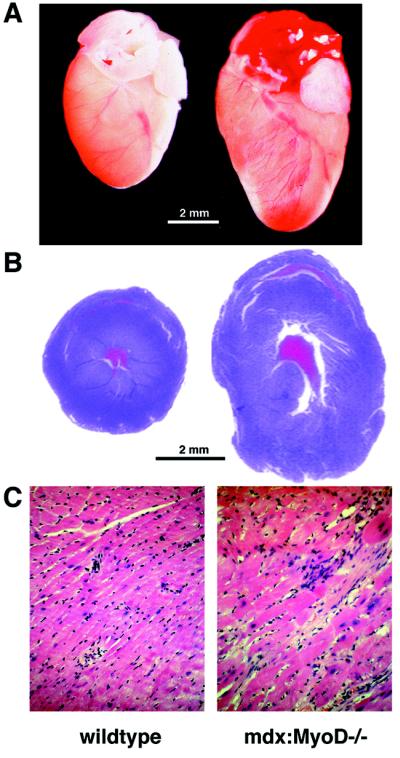
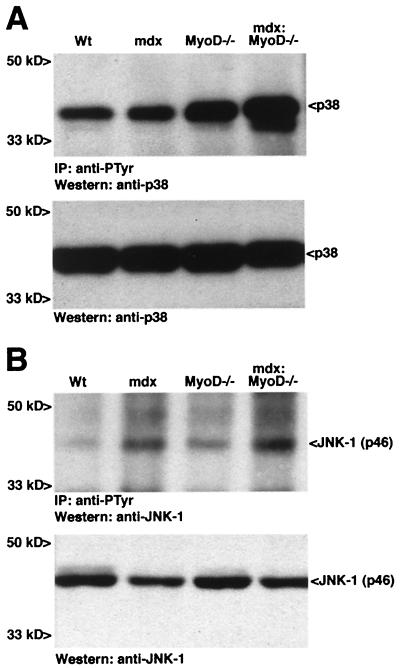
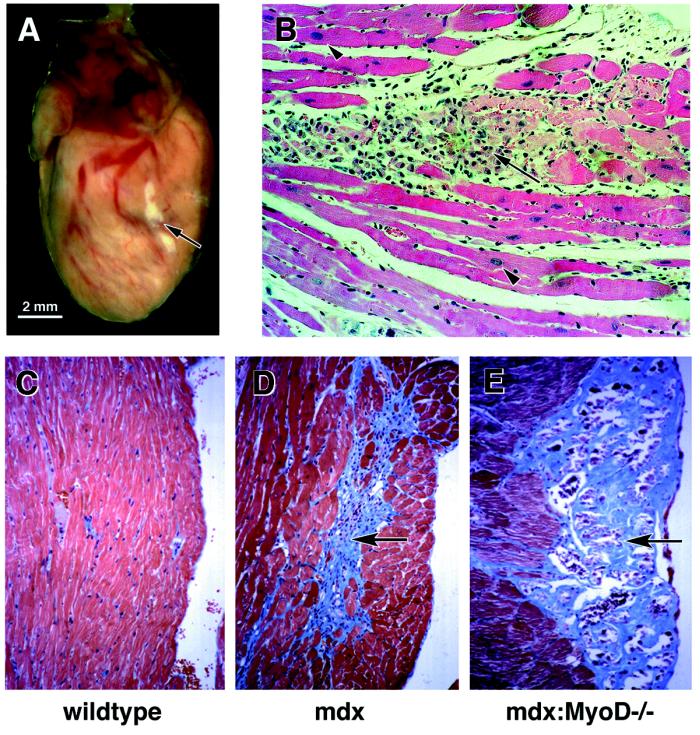
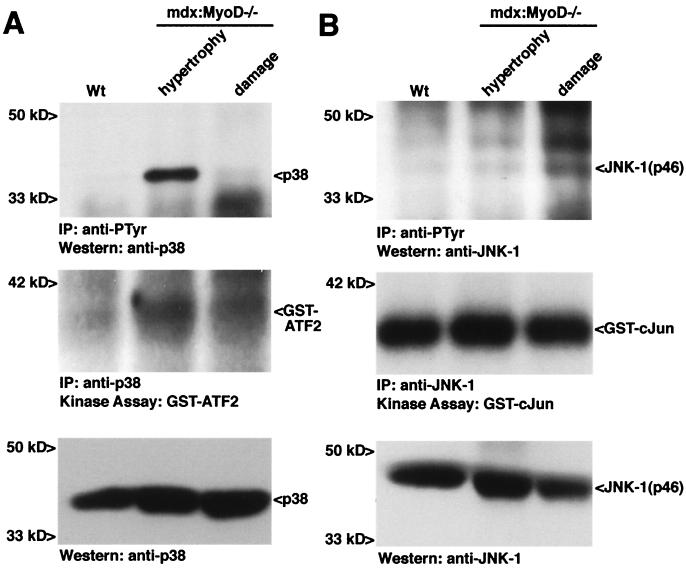
The mdx mouse, a mouse model of Duchenne muscular dystrophy, carries a loss-of-function mutation in dystrophin, a component of the membrane-associated dystrophin-glycoprotein complex. Unlike humans, mdx mice rarely display cardiac abnormalities and exhibit dystrophic changes only in a small number of heavily used skeletal muscle groups. By contrast, mdx:MyoD-/- mice lacking dystrophin and the skeletal muscle-specific bHLH transcription factor MyoD display a severe skeletal myopathy leading to widespread dystrophic changes in skeletal muscle and premature death around 1 year of age. The severely increased phenotype of mdx:MyoD-/- muscle is a consequence of impaired muscle regeneration caused by enhanced satellite cell self-renewal. Here we report that mdx:MyoD-/- mice developed a severe cardiac myopathy with areas of necrosis associated with hypertrophied myocytes. Moreover, heart tissue from mdx:MyoD-/- mice exhibited constitutive activation of stress-activated signaling components, similar to in vitro models of cardiac myocyte adaptation. Taken together, these results support the hypothesis that the progression of skeletal muscle damage is a significant contributing factor leading to development of cardiomyopathy.
Figures
Similar articles
-
Regeneration and myogenic cell proliferation correlate with taurine levels in dystrophin- and MyoD-deficient muscles.Anat Rec. 1998 Oct;252(2):311-24. doi: 10.1002/(SICI)1097-0185(199810)252:2<311::AID-AR17>3.0.CO;2-Q. Anat Rec. 1998. PMID: 9776086
-
Unacylated Ghrelin Enhances Satellite Cell Function and Relieves the Dystrophic Phenotype in Duchenne Muscular Dystrophy mdx Model.Stem Cells. 2017 Jul;35(7):1733-1746. doi: 10.1002/stem.2632. Epub 2017 May 7. Stem Cells. 2017. PMID: 28436144
-
MyoD is required for myogenic stem cell function in adult skeletal muscle.Genes Dev. 1996 May 15;10(10):1173-83. doi: 10.1101/gad.10.10.1173. Genes Dev. 1996. PMID: 8675005
-
Proteomic profiling of the dystrophin-deficient mdx phenocopy of dystrophinopathy-associated cardiomyopathy.Biomed Res Int. 2014;2014:246195. doi: 10.1155/2014/246195. Epub 2014 Mar 20. Biomed Res Int. 2014. PMID: 24772416 Free PMC article. Review.
-
Cardiomyopathy in animal models of muscular dystrophy.Curr Opin Cardiol. 2001 May;16(3):211-7. doi: 10.1097/00001573-200105000-00009. Curr Opin Cardiol. 2001. PMID: 11357018 Review.
Cited by
-
Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart.Circulation. 2003 Sep 30;108(13):1626-32. doi: 10.1161/01.CIR.0000089371.11664.27. Epub 2003 Sep 2. Circulation. 2003. PMID: 12952841 Free PMC article.
-
Role of the Renin-Angiotensin-Aldosterone System in Dystrophin-Deficient Cardiomyopathy.Int J Mol Sci. 2020 Dec 31;22(1):356. doi: 10.3390/ijms22010356. Int J Mol Sci. 2020. PMID: 33396334 Free PMC article. Review.
-
Ablation of cyclase-associated protein 2 (CAP2) leads to cardiomyopathy.Cell Mol Life Sci. 2013 Feb;70(3):527-43. doi: 10.1007/s00018-012-1142-y. Epub 2012 Sep 4. Cell Mol Life Sci. 2013. PMID: 22945801 Free PMC article.
-
Single SERCA2a Therapy Ameliorated Dilated Cardiomyopathy for 18 Months in a Mouse Model of Duchenne Muscular Dystrophy.Mol Ther. 2020 Mar 4;28(3):845-854. doi: 10.1016/j.ymthe.2019.12.011. Epub 2020 Jan 10. Mol Ther. 2020. PMID: 31981493 Free PMC article.
-
Identification of gene co-regulatory modules and associated cis-elements involved in degenerative heart disease.BMC Med Genomics. 2009 May 28;2:31. doi: 10.1186/1755-8794-2-31. BMC Med Genomics. 2009. PMID: 19476647 Free PMC article.
References
-
- Emery A E H. Duchenne Muscular Dystrophy. 2nd Ed. Oxford: Oxford Univ. Press; 1993.
-
- Frankel K A, Rosser R J. Hum Pathol. 1976;7:375–386. - PubMed
-
- Nigro G, Comi L, Politano L, Bain R J I. Int J Cardiol. 1990;26:271–277. - PubMed
-
- Politano L, Nigro V, Nigro G, Petretta V R, Passamano L, Papparella S, DiSomma S, Comi L I. J Am Med Assoc. 1996;275:1335–1338. - PubMed
-
- Cox G F, Kunkel L M. Curr Opin Cardiol. 1997;12:329–343. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
