

The evolutionary biology and population genetics underlying fungal strain typing
- PMID: 9880478
- PMCID: PMC88910
- DOI: 10.1128/CMR.12.1.126
The evolutionary biology and population genetics underlying fungal strain typing
Abstract
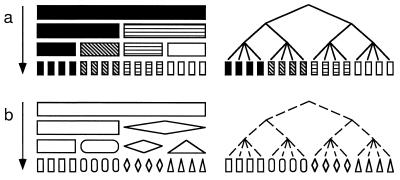
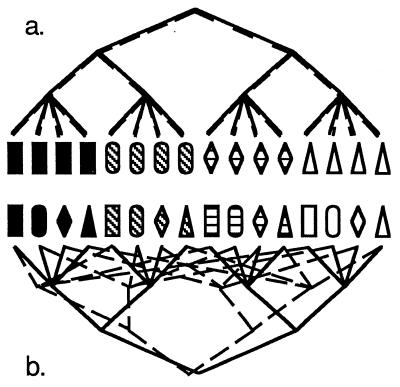
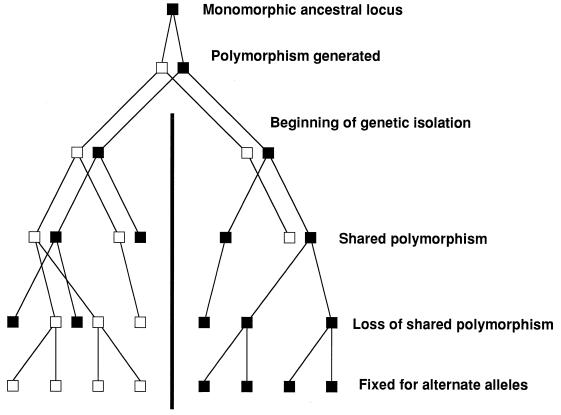
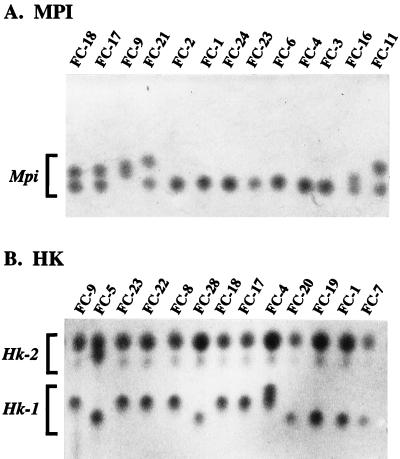
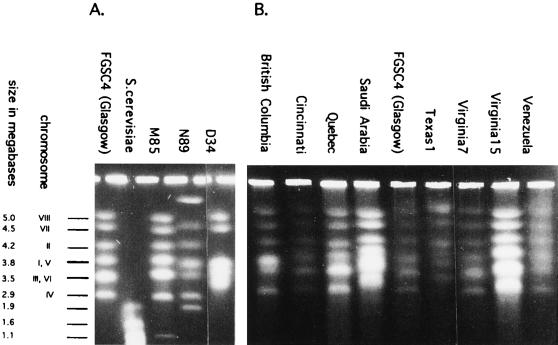
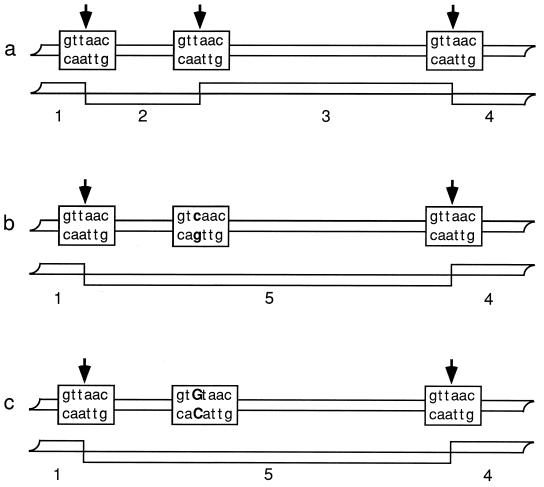
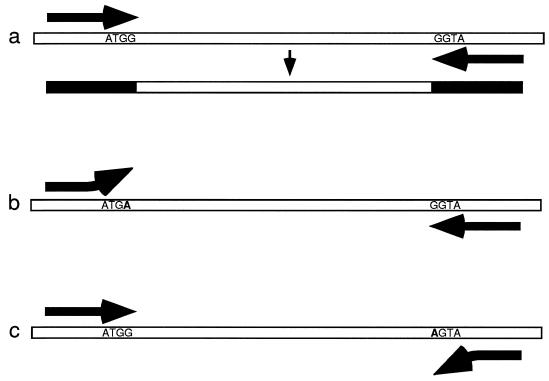
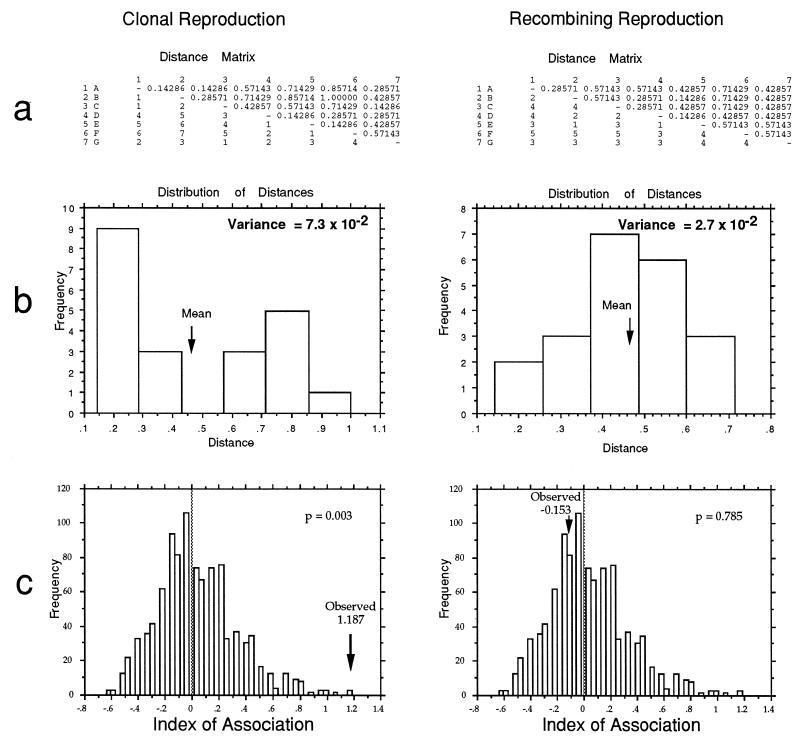
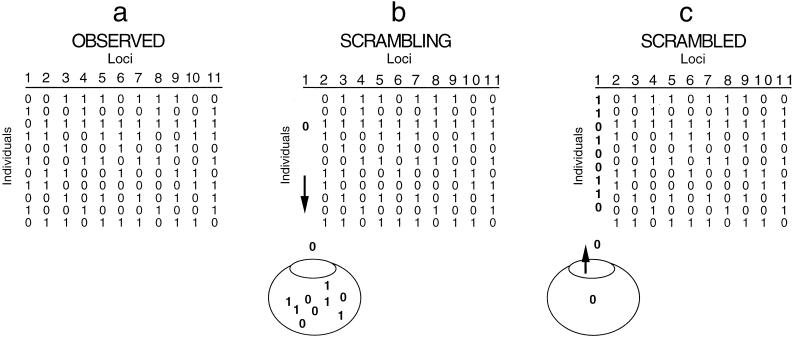
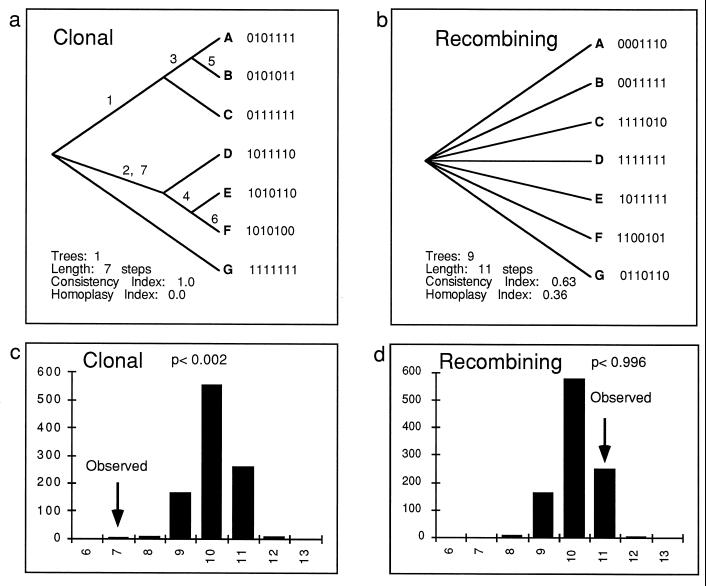
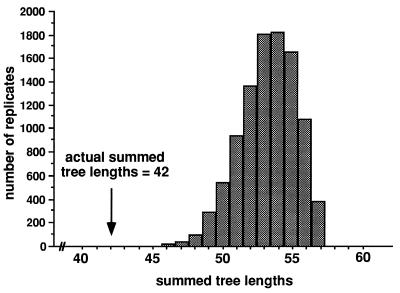
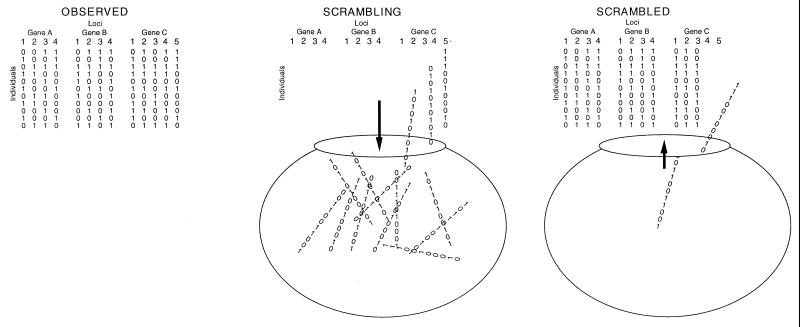
Strain typing of medically important fungi and fungal population genetics have been stimulated by new methods of tapping DNA variation. The aim of this contribution is to show how awareness of fungal population genetics can increase the utility of strain typing to better serve the interests of medical mycology. Knowing two basic features of fungal population biology, the mode of reproduction and genetic differentiation or isolation, can give medical mycologists information about the intraspecific groups that are worth identifying and the number and type of markers that would be needed to do so. The same evolutionary information can be just as valuable for the selection of fungi for development and testing of pharmaceuticals or vaccines. The many methods of analyzing DNA variation are evaluated in light of the need for polymorphic loci that are well characterized, simple, independent, and stable. Traditional population genetic and new phylogenetic methods for analyzing mode of reproduction, genetic differentiation, and isolation are reviewed. Strain typing and population genetic reports are examined for six medically important species: Coccidioides immitis, Histoplasma capsulatum, Candida albicans, Cryptococcus neoformans, Aspergillus fumigatus, and A. flavus. Research opportunities in the areas of genomics, correlation of clinical variation with genetic variation, amount of recombination, and standardization of approach are suggested.
Figures

References
-
- Anderson J B, Kohn L M. Clonality in soilborne, plant-pathogenic fungi. Annu Rev Phytopathol. 1995;33:369–391. - PubMed
-
- Anderson J B, Korhonen K, Ullrich R C. Relationships between European and North American biological species of Armillaria mellea. Exp Mycol. 1980;4:87–95.
-
- Appel D J, Gordon T R. Relationships among pathogenic and nonpathogenic isolates of Fusarium oxysporum based on the partial sequence of the intergenic spacer region of the ribosomal DNA. Mol Plant-Microbe Interact. 1996;9:125–138. - PubMed
-
- Archie J W. A randomization test for phylogenetic information in systematic data. Syst Zool. 1989;38:239–252.
-
- Aulakh H S, Strauss S E, Kwon-Chung K J. Genetic relatedness of Filobasidiella neoformans (Cryptococcus neoformans) and Filobasidiella bacillispora (Cryptococcus bacillosporus) as determined by deoxyribonucleic acid base composition and sequence homology studies. Int J Syst Bacteriol. 1981;31:97–103.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
