

Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1
- PMID: 9887100
- PMCID: PMC316375
- DOI: 10.1101/gad.13.1.64
Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1
Abstract
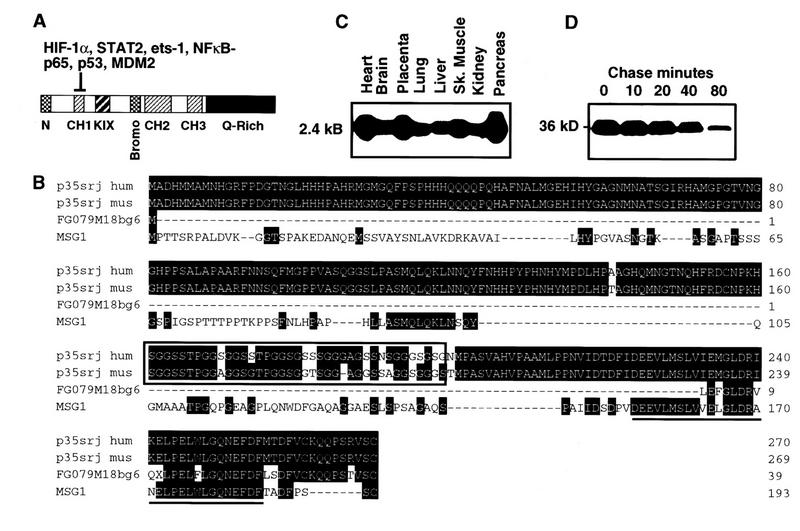
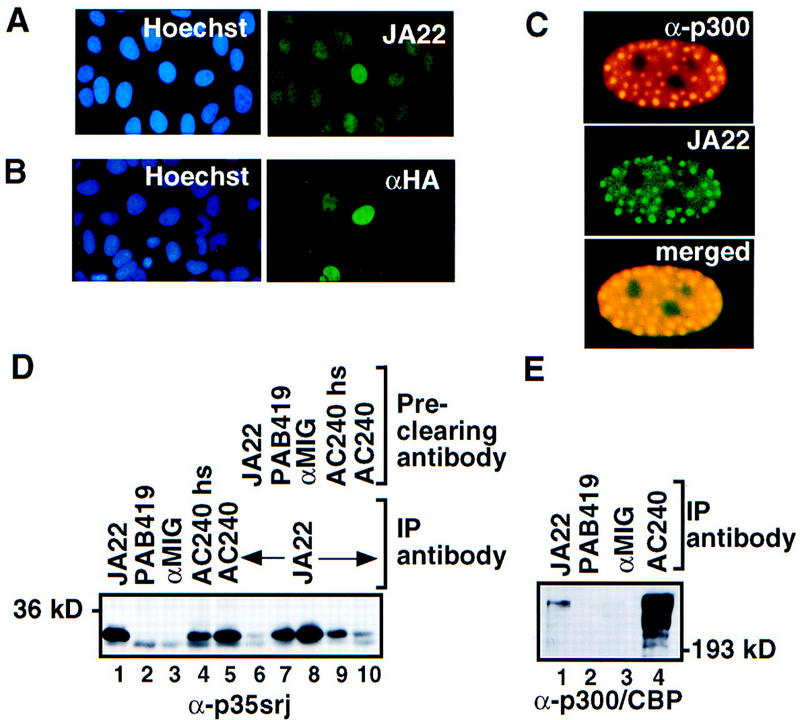
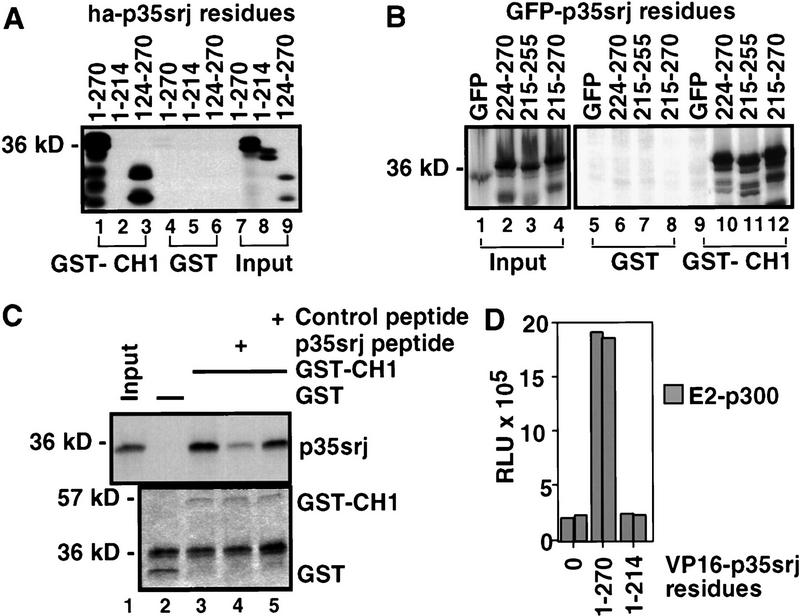
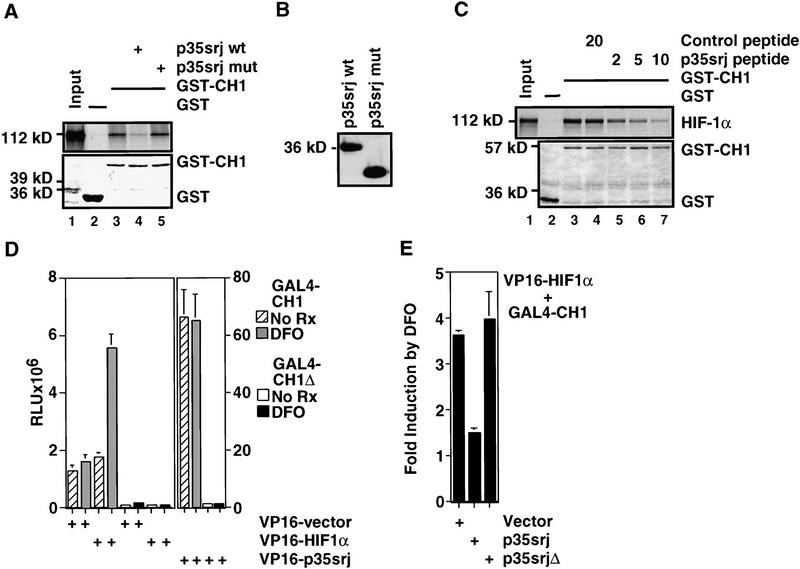
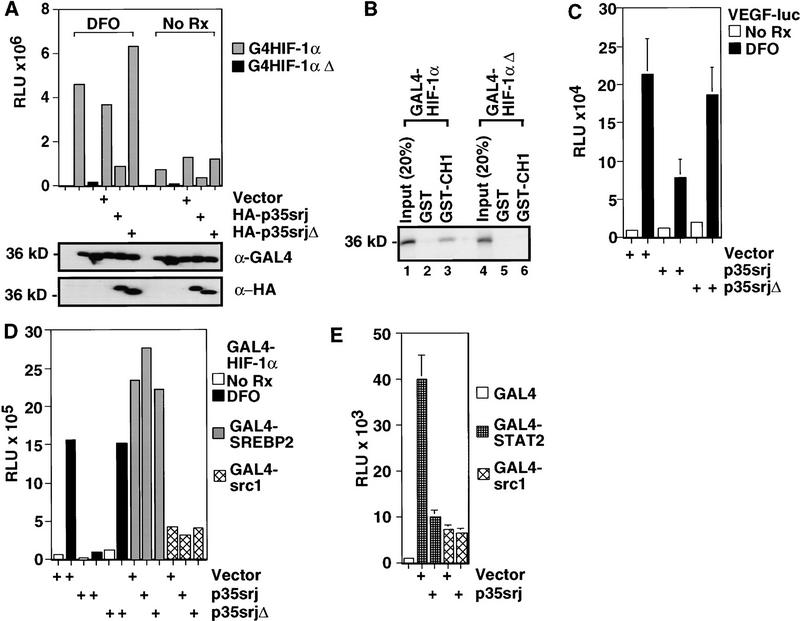
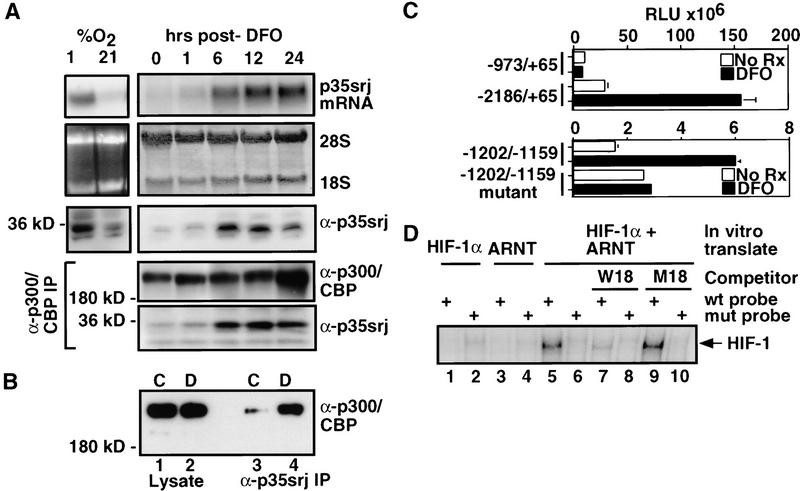
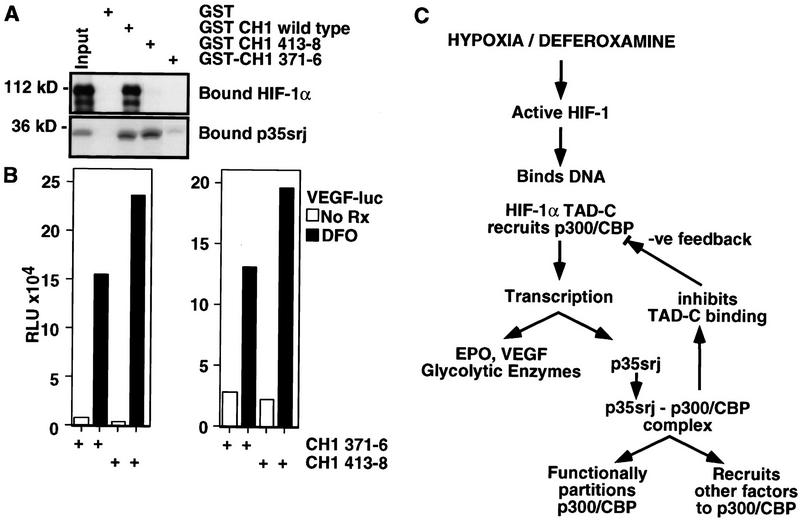
Recruitment of p300/CBP by the hypoxia-inducible factor, HIF-1, is essential for the transcriptional response to hypoxia and requires an interaction between the p300/CBP CH1 region and HIF-1alpha. A new p300-CH1 interacting protein, p35srj, has been identified and cloned. p35srj is an alternatively spliced isoform of MRG1, a human protein of unknown function. Virtually all endogenous p35srj is bound to p300/CBP in vivo, and it inhibits HIF-1 transactivation by blocking the HIF-1alpha/p300 CH1 interaction. p35srj did not affect transactivation by transcription factors that bind p300/CBP outside the CH1 region. Endogenous p35srj is up-regulated markedly by the HIF-1 activators hypoxia or deferoxamine, suggesting that it could operate in a negative-feedback loop. In keeping with this notion, a p300 CH1 mutant domain, defective in HIF-1 but not p35srj binding, enhanced endogenous HIF-1 function. In hypoxic cells, p35srj may regulate HIF-1 transactivation by controlling access of HIF-1alpha to p300/CBP, and may keep a significant portion of p300/CBP available for interaction with other transcription factors by partially sequestering and functionally compartmentalizing cellular p300/CBP.
Figures
References
-
- Arany Z, Newsome D, Oldread E, Livingston DM, Eckner R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature. 1995;374:81–84. - PubMed
-
- Ausubel F, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short protocols in molecular biology. New York, NY: John Wiley & Sons; 1995.
-
- Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. - PubMed
-
- Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D’Andrea A, Livingston DM. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-α. Nature. 1996;383:344–347. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous