

Complementation of defective colony-stimulating factor 1 receptor signaling and mitogenesis by Raf and v-Src
- PMID: 9891045
- PMCID: PMC116040
- DOI: 10.1128/MCB.19.2.1101
Complementation of defective colony-stimulating factor 1 receptor signaling and mitogenesis by Raf and v-Src
Abstract
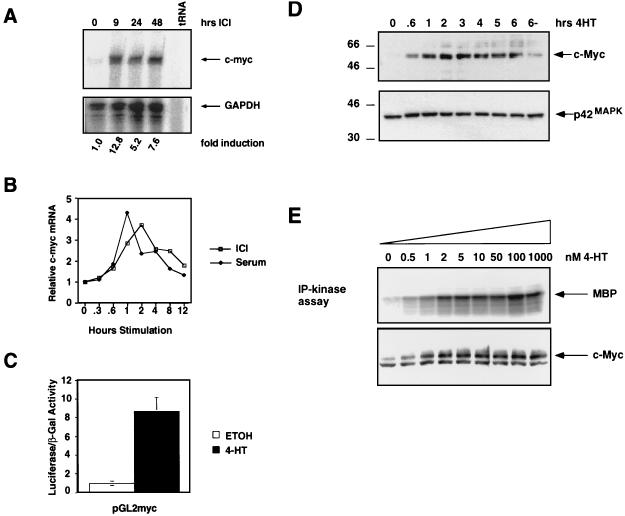
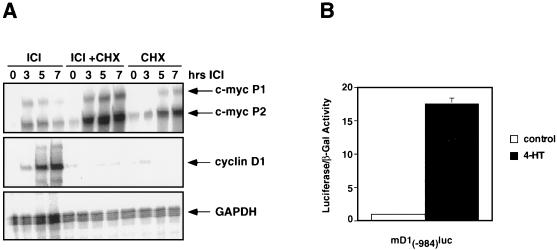
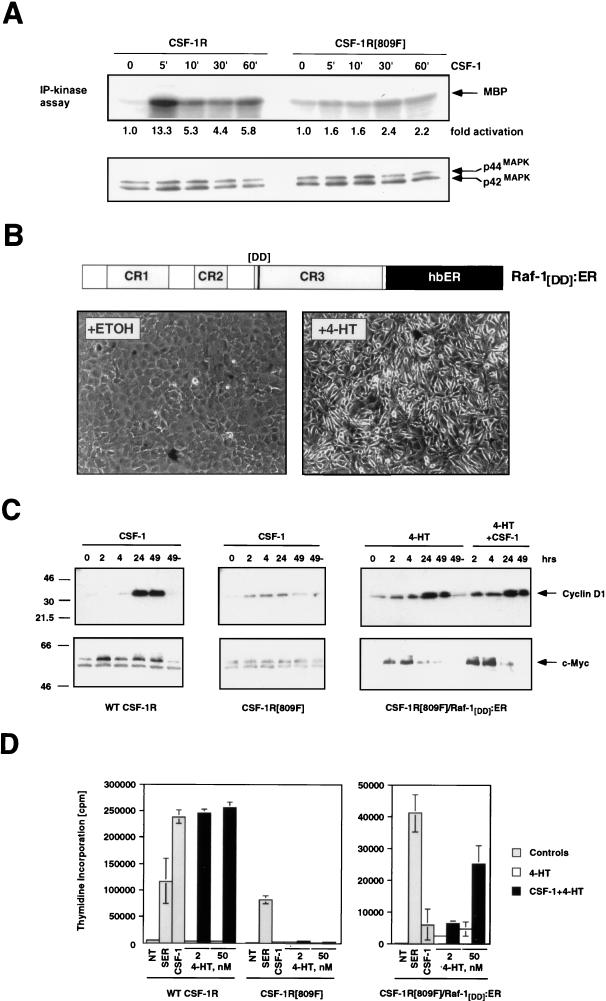
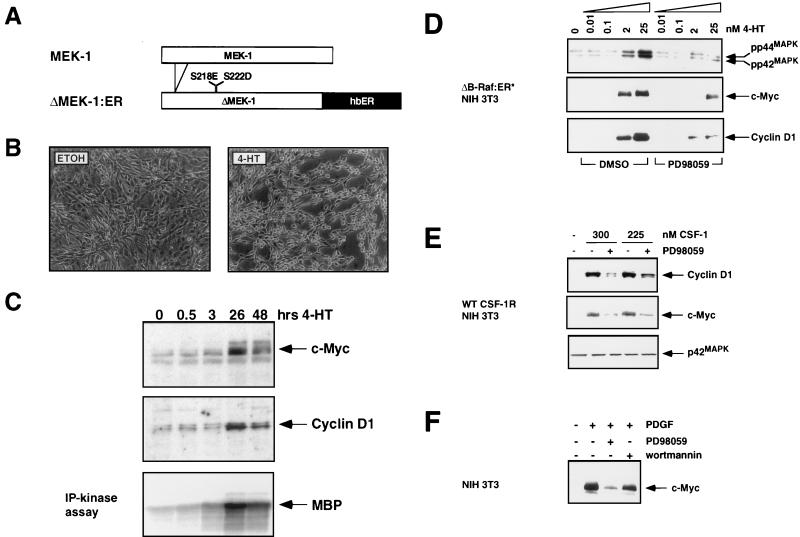
Ras-activated signal transduction pathways are implicated in the control of cell proliferation, differentiation, apoptosis, and tumorigenesis, but the molecular mechanisms mediating these diverse functions have yet to be fully elucidated. Conditionally active forms of Raf, v-Src, and MEK1 were used to identify changes in gene expression that participate in oncogenic transformation, as well as in normal growth control. Activation of Raf, v-Src, and MEK1 led to induced expression of c-Myc and cyclin D1. Induction of c-Myc mRNA by Raf was an immediate-early response, whereas the induction of cyclin D1 mRNA was delayed and inhibited by cycloheximide. Raf activation also resulted in the induction of an established c-Myc target gene, ornithine decarboxylase (ODC). ODC induction by Raf was mediated, in part, by tandem E-boxes contained in the first intron of the gene. Activation of the human colony-stimulating factor 1 (CSF-1) receptor in NIH 3T3 cells leads to activation of the mitogen-activated protein (MAP) kinase pathway and induced expression of c-Fos, c-Myc, and cyclin D1, leading to a potent mitogenic response. By contrast, a mutated form of this receptor fails to activate the MAP kinases or induce c-Myc and cyclin D1 expression and fails to elicit a mitogenic response. The biological significance of c-Myc and cyclin D1 induction by Raf and v-Src was confirmed by the demonstration that both of these protein kinases complemented the signaling and mitogenic defects of cells expressing this mutated form of the human CSF-1 receptor. Furthermore, the induction of c-Myc and cyclin D1 by oncogenes and growth factors was inhibited by PD098059, a specific MAP kinase kinase (MEK) inhibitor. These data suggest that the Raf/MEK/MAP kinase pathway plays an important role in the regulation of c-Myc and cyclin D1 expression in NIH 3T3 cells. The ability of oncogenes such as Raf and v-Src to regulate the expression of these proteins reveals new lines of communication between cytosolic signal transducers and the cell cycle machinery.
Figures

References
-
- Abrahamsen M S, Li R S, Dietrich-Goetz W, Morris D R. Multiple DNA elements responsible for transcriptional regulation of the ornithine decarboxylase gene by protein kinase A. J Biol Chem. 1992;267:18866–18873. - PubMed
-
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell R G. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. - PubMed
-
- Alberts A S, Geneste O, Treisman R. Activation of SRF-regulated chromosomal templates by Rho-family GTPases requires a signal that also induces H4 hyperacetylation. Cell. 1998;92:475–487. - PubMed
-
- Alessi D R, Cuenda A, Cohen P, Dudley D T, Saltiel A R. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. - PubMed
-
- Amati B, Land H. Myc-Max-Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Curr Opin Genet Dev. 1994;4:102–108. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous