

Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways
- PMID: 9916130
- PMCID: PMC407875
- DOI: 10.1172/JCI3326
Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways
Abstract
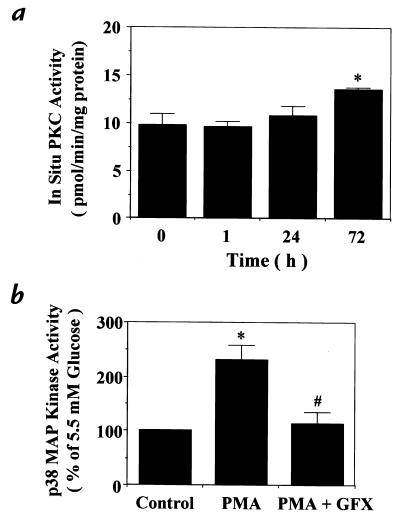
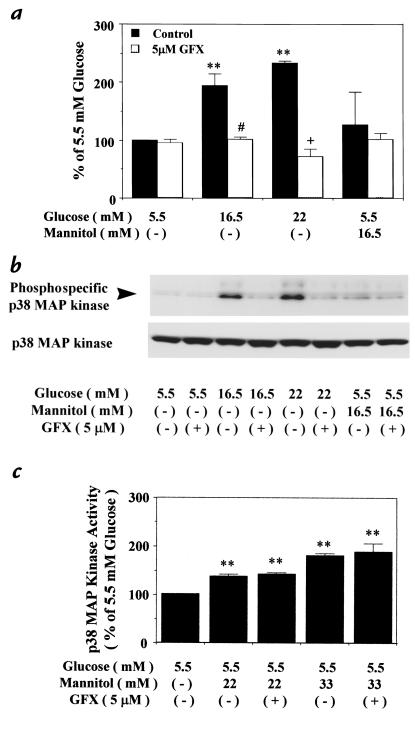
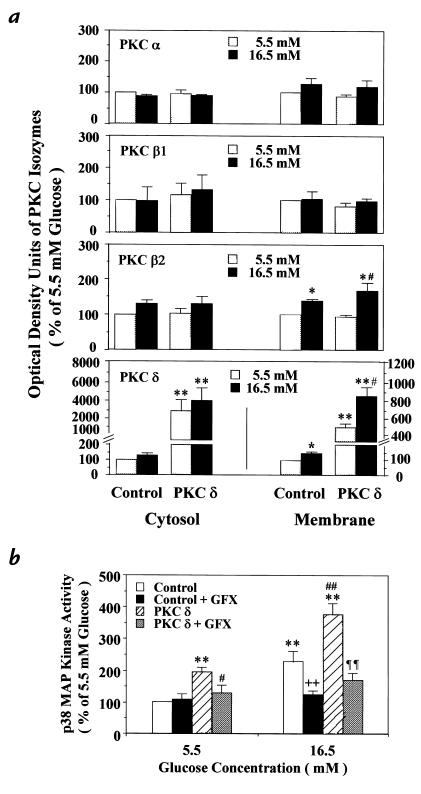
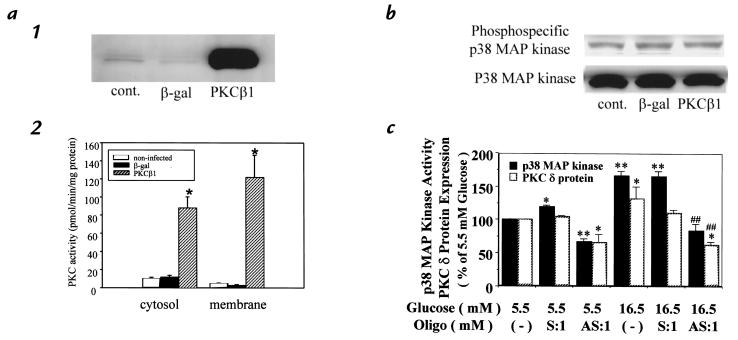
Hyperglycemia can cause vascular dysfunctions by multiple factors including hyperosmolarity, oxidant formation, and protein kinase C (PKC) activation. We have characterized the effect of hyperglycemia on p38 mitogen-activated protein (p38) kinase activation, which can be induced by oxidants, hyperosmolarity, and proinflammatory cytokines, leading to apoptosis, cell growth, and gene regulation. Glucose at 16.5 mM increased p38 kinase activity in a time-dependent manner compared with 5.5 mM in rat aortic smooth muscle cells (SMC). Mannitol activated p38 kinase only at or greater than 22 mM. High glucose levels and a PKC agonist activated p38 kinase, and a PKC inhibitor, GF109203X, prevented its activation. However, p38 kinase activation by mannitol or tumor necrosis factor-alpha was not inhibited by GF109203X. Changes in PKC isoform distribution after exposure to 16.5 mM glucose in SMC suggested that both PKC-beta2 and PKC-delta isoforms were increased. Activities of p38 kinase in PKC-delta- but not PKC-beta1-overexpressed SMC were increased compared with control cells. Activation of p38 kinase was also observed and characterized in various vascular cells in culture and aorta from diabetic rats. Thus, moderate hyperglycemia can activate p38 kinase by a PKC-delta isoform-dependent pathway, but glucose at extremely elevated levels can also activate p38 kinase by hyperosmolarity via a PKC-independent pathway.
Figures



References
-
- The Diabetes ControlComplications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. - PubMed
-
- Greene D, Lattimer SA, Sima AAF. Sorbitol, phosphoinositides and sodium-potassium-ATPase in the pathogenesis of diabetic complications. N Engl J Med. 1987;316:599–606. - PubMed
-
- Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–1321. - PubMed
-
- Williamson JR, et al. Hyperglycemic pseudhypoxia and diabetic complications. Diabetes. 1993;42:801–813. - PubMed
-
- Koya D, King GL. Protein kinase C activation and the development of diabetic complication. Diabetes. 1998;47:859–866. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
