

Review
doi: 10.1172/JCI6277.
The innate immune system in cystic fibrosis lung disease
Affiliations
- PMID: 9927489
- PMCID: PMC407907
- DOI: 10.1172/JCI6277
Item in Clipboard
Review
The innate immune system in cystic fibrosis lung disease
J Clin Invest.
1999 Feb.
No abstract available
Figures
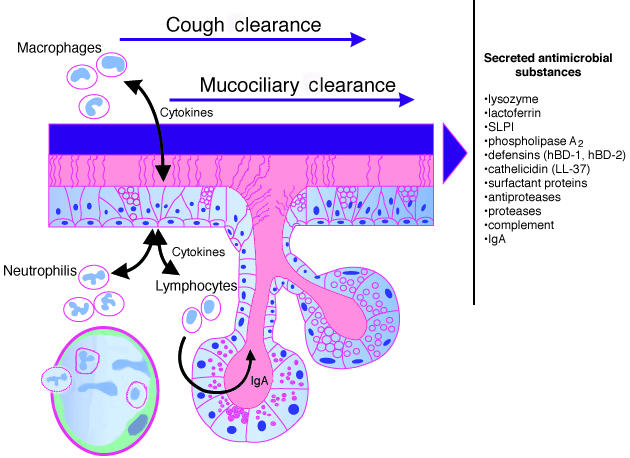
Host defense mechanisms of the respiratory epithelium. Cough and cilia mechanically remove inhaled debris and microorganisms entrapped in mucus, a mechanism called mucociliary clearance. Multiple substances with pro- and antiinflammatory as well as antimicrobial activities are secreted by epithelial cells and function as an innate immune barrier. Macrophages, B and T lymphocytes, neutrophils and also epithelial cells represent cellular components of the innate and adaptive immune systems.
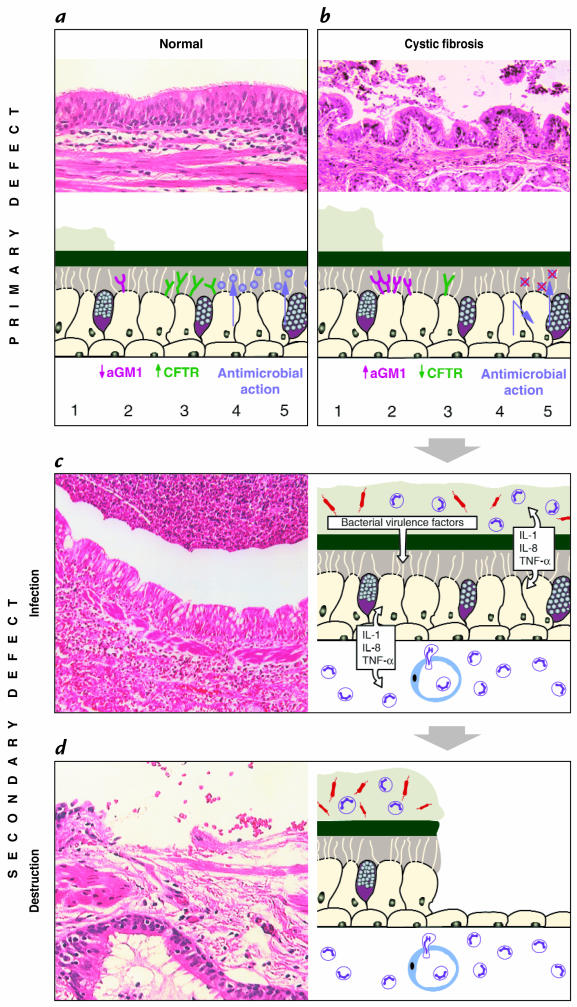
Schematic overview of the development of lung disease in CF. A diagram explaining the molecular mechanisms is accompanied by micrographs of representative lung tissue. Several hypotheses describe the link between defects in CFTR and increased susceptibility to bacterial infection. Panel a and b compare normal and CF airway epithelium. As compared with the situation in normal lungs (a), in CF, (b) defects of CFTR might result in dehydrated secretions, resulting in decreased clearance and subsequent colonization with pathogens (1). CF epithelia may express more binding receptors for Pseudomonas aeruginosa, such as asialoGM1 (aGM1) (2), resulting in increased adhesion of this pathogen. CFTR present on the apical surface of airway cells might function as a cellular scavenger receptor for P. aeruginosa in order to clear the bacterium from the airway lumen (3). Antimicrobial substances are secreted (4, 5) into the aqueous sol layer and exhibit local antibacterial activity. In CF, secretion of antimicrobials might be impaired (4), or secreted substances can be inactivated by the CF-specific milieu, such as high salt concentration (5). The primary defect in host defense results in infection with accompanying inflammation. The chronic inflammation causes secondary defects of the host defense system. (c) Chronic infection of CF airways. Numerous bacterial organisms and neutrophils are present in the thick layer of mucus. Inflammatory and epithelial cells secrete multiple proinflammatory cytokines as well as proteases. Bacteria release proteases, LPS, and other virulence factors. An intense neutrophilic infiltrate is also found within the interstitium of the epithelium. (d) The characteristic tissue destruction in later stages of the disease. Shedding of the epithelium has taken place, and the remaining epithelium has undergone metaplastic changes. The intense inflammatory response persists and remains acute in nature, rather than the expected change to a lymphocytic response. CF, cystic fibrosis; CFTR, CF transmembrane conductance regulator, LPS, lipopolysaccharides.
Comment in
-
Status of gene therapy for cystic fibrosis lung disease.J Clin Invest. 1999 Feb;103(4):441-5. doi: 10.1172/JCI6330. J Clin Invest. 1999. PMID: 10021450 Free PMC article. Review. No abstract available.
References
-
- Quinton P. Chloride impermeability in cystic fibrosis. Nature. 1983;301:421–422. - PubMed
-
- Rommens J, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. - PubMed
-
- Riordan J, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Smith J, Travis S, Greenberg E, Welsh M. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85:229–236. - PubMed
