

Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex
- PMID: 9971795
- PMCID: PMC104457
- DOI: 10.1128/JVI.73.3.2126-2135.1999
Human immunodeficiency virus type 1 integrase protein promotes reverse transcription through specific interactions with the nucleoprotein reverse transcription complex
Abstract
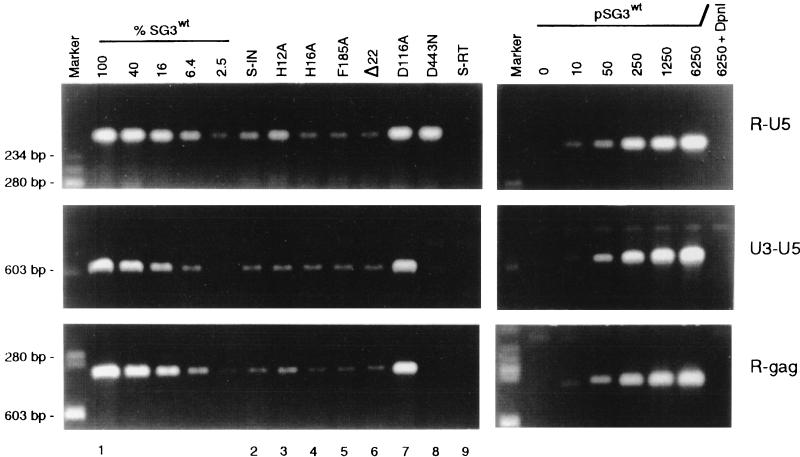
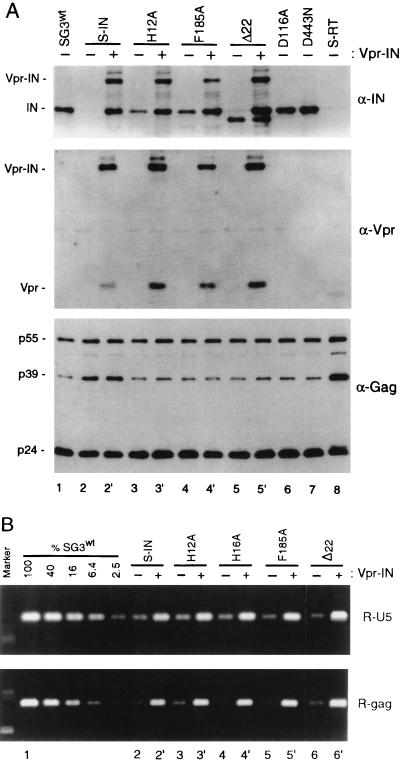
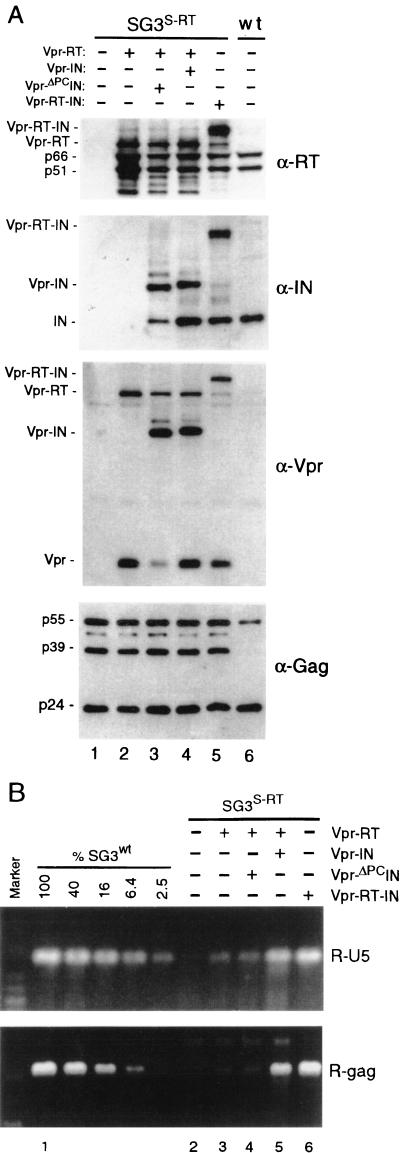
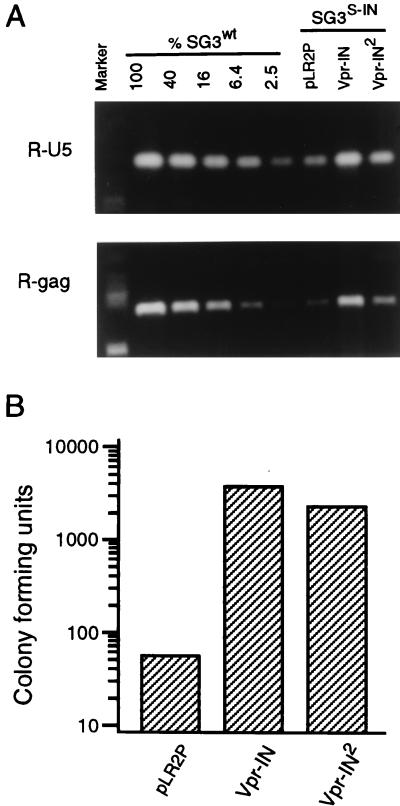
The human immunodeficiency virus type 1 (HIV-1) integrase protein (IN) is essential for integration of the viral DNA into host cell chromosomes. Since IN is expressed and assembled into virions as part of the 160-kDa Gag-Pol precursor polyprotein and catalyzes integration of the provirus in infected cells as a mature 32-kDa protein, mutations in IN are pleiotropic and may affect virus replication at different stages of the virus life cycle in addition to integration. Several different phenotypes have been observed for IN mutant viruses, including defects in virion morphology, protein composition, reverse transcription, nuclear import, and integration. Because the effects of mutations in the IN domain of Gag-Pol can not always be distinguished from those of mutations in the mature IN protein, there remains a significant gap in our understanding of IN function in vivo. To directly analyze the function of the mature IN protein itself, in the context of a replicating virus but independently from that of Gag-Pol, we used an approach developed in our laboratory for incorporating proteins into HIV virions by their expression in trans as fusion partners of either Vpr or Vpx. By providing IN in trans as a Vpr-IN fusion protein, our analysis revealed, for the first time, that the mature IN protein is essential for the efficient initiation of reverse transcription in infected cells and that this function does not require the IN protein to be enzymatically (integration) active. Our findings of a direct physical interaction between IN and reverse transcriptase and the failure of heterologous HIV-2 IN protein to efficiently support reverse transcription indicate that this novel function occurs through specific interactions with other viral components of the reverse transcription initiation complex. Studies involving complementation between integration- and DNA synthesis-defective IN mutants further support this conclusion and reveal that the highly conserved HHCC motif of IN is important for both activities. These findings provide important new insights into IN function and reverse transcription in the context of the nucleoprotein reverse transcription complex within the infected cell. Moreover, they validate a novel approach that obviates the need to mutate Gag-Pol in order to study the role of its individual mature components at the virus replication level.
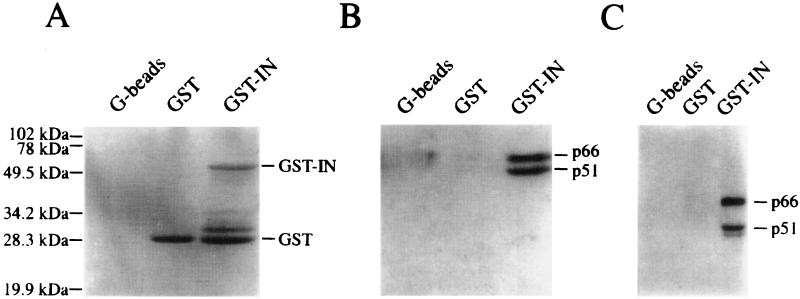
Figures

References
-
- Brown P. Integration. In: Coffin J M, Hughes S H, Varmus H E, editors. Retroviruses. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1997. pp. 161–204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
