

Amino-terminal sequences of sigmaN (sigma54) inhibit RNA polymerase isomerization
- PMID: 9990859
- PMCID: PMC316430
- DOI: 10.1101/gad.13.3.357
Amino-terminal sequences of sigmaN (sigma54) inhibit RNA polymerase isomerization
Abstract
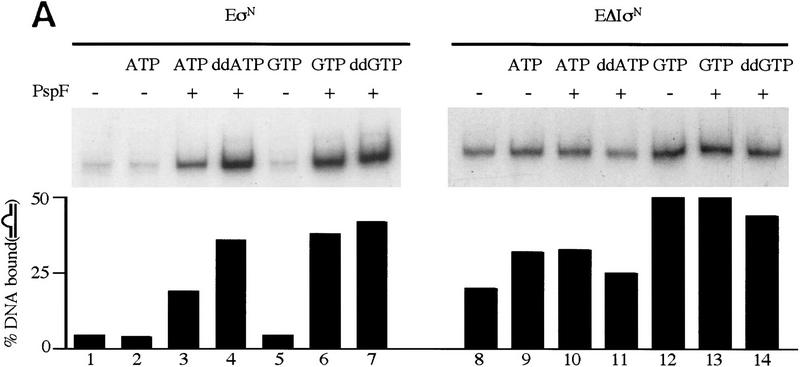
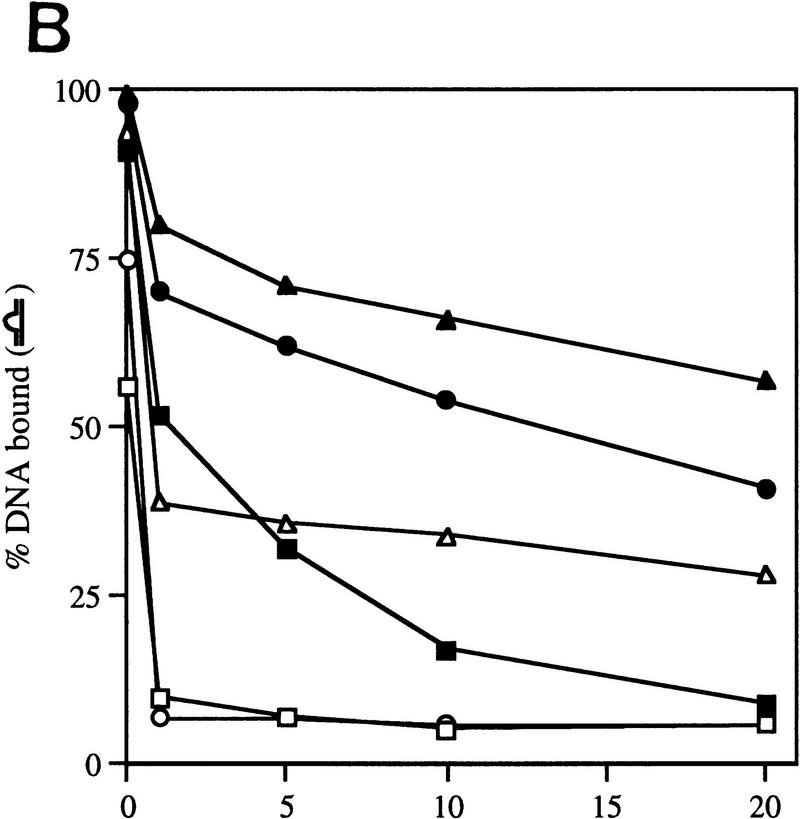
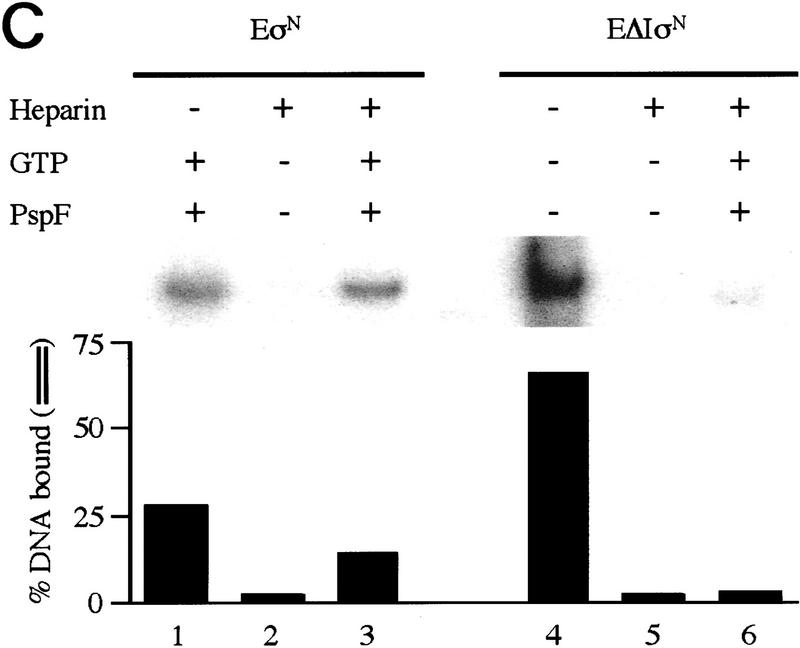
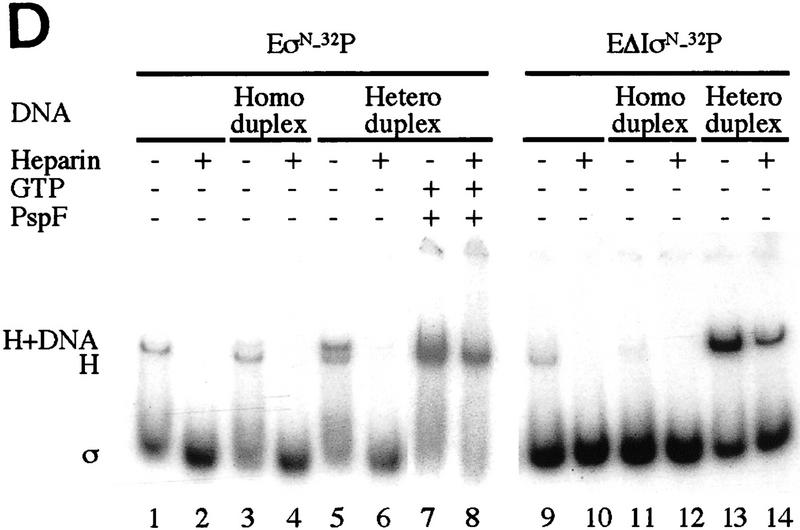
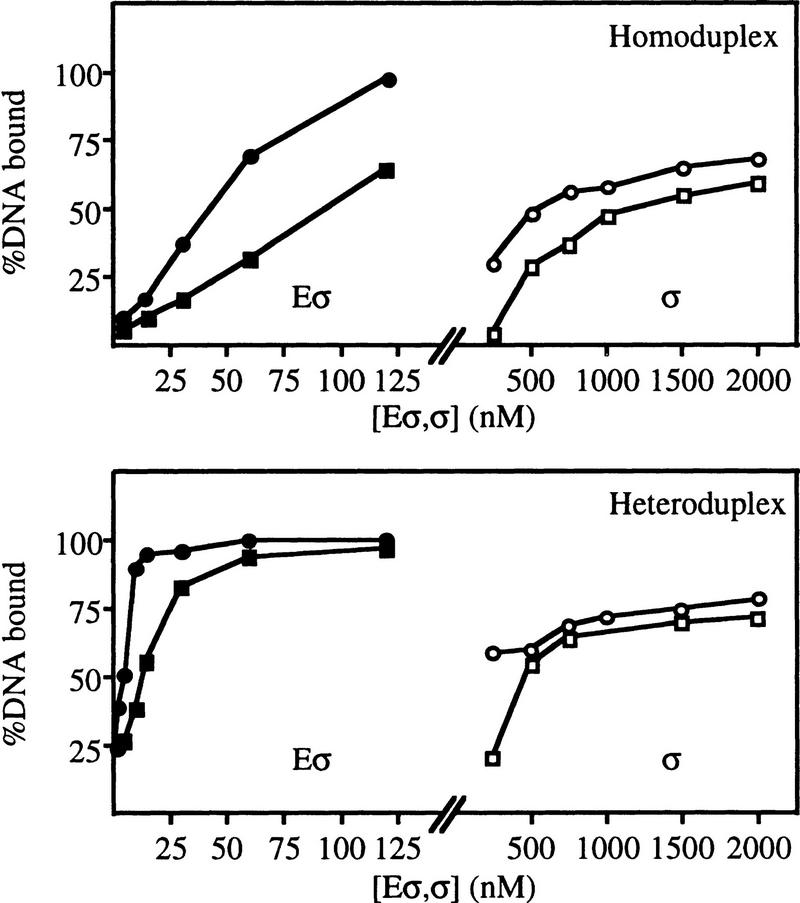
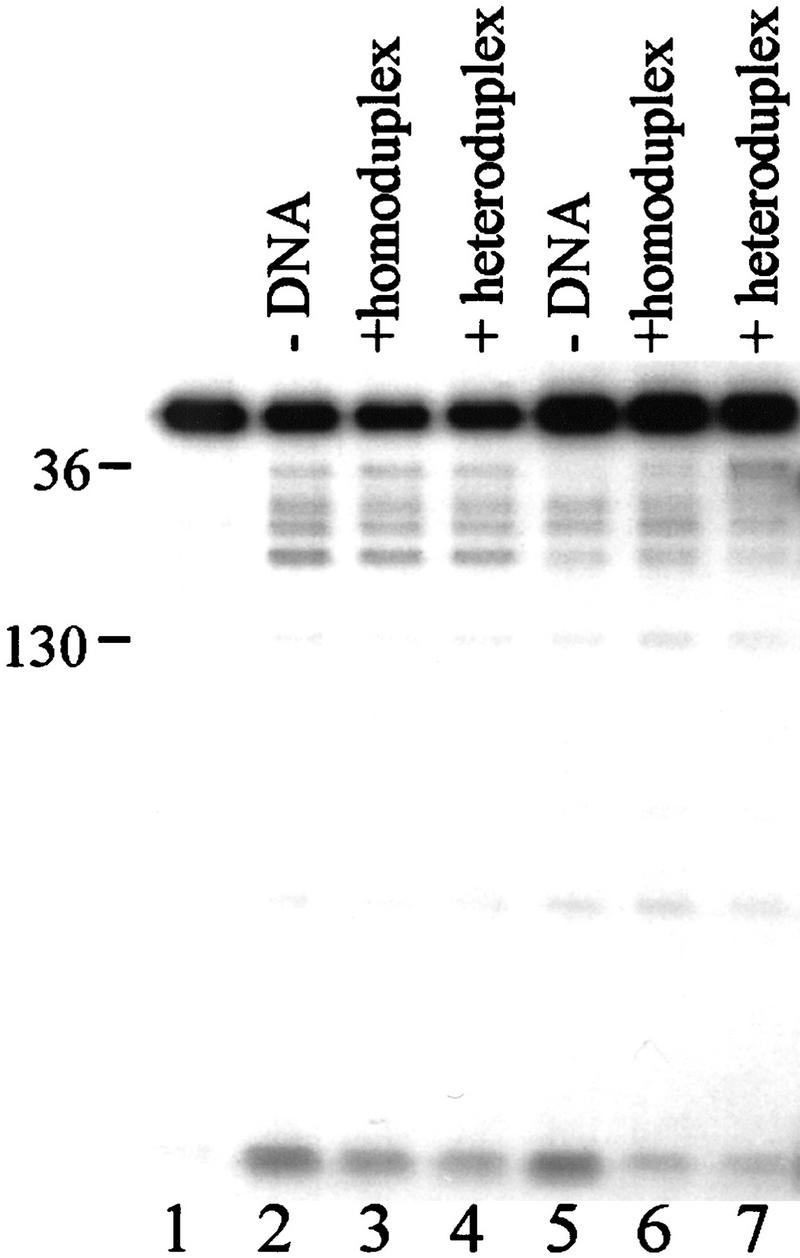
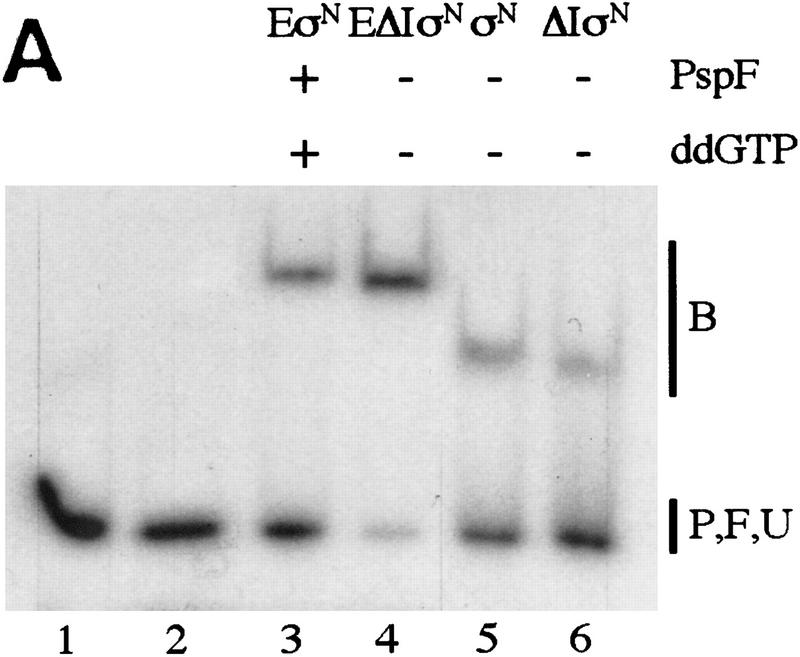
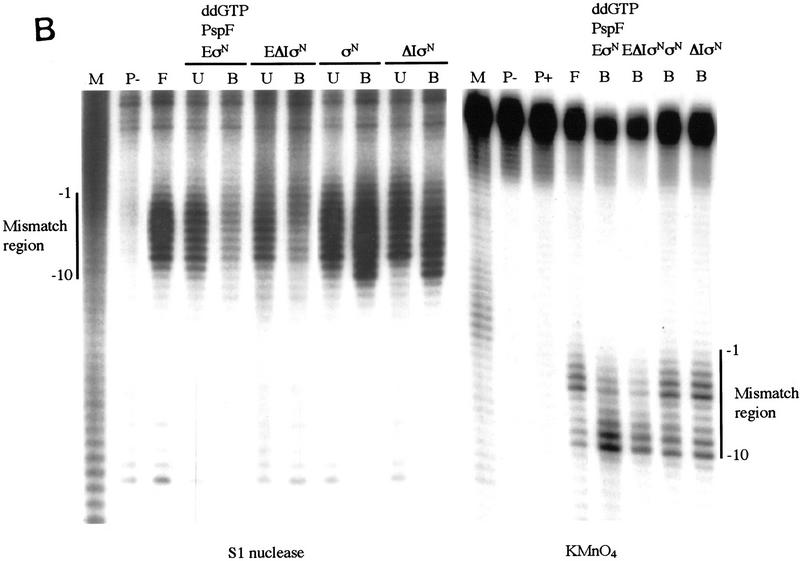
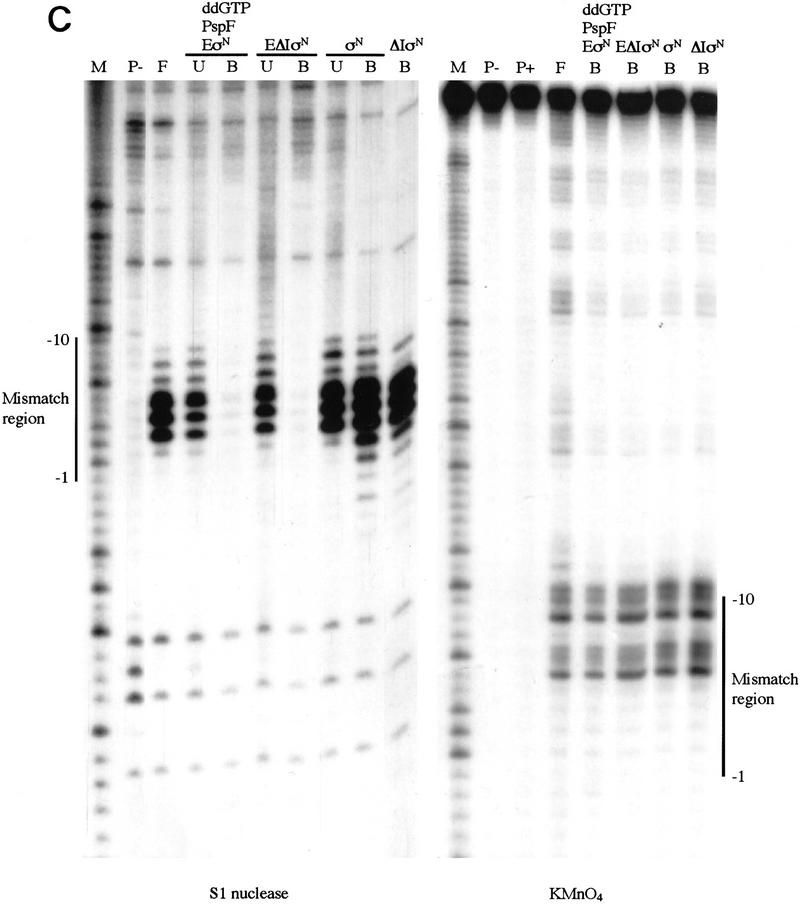
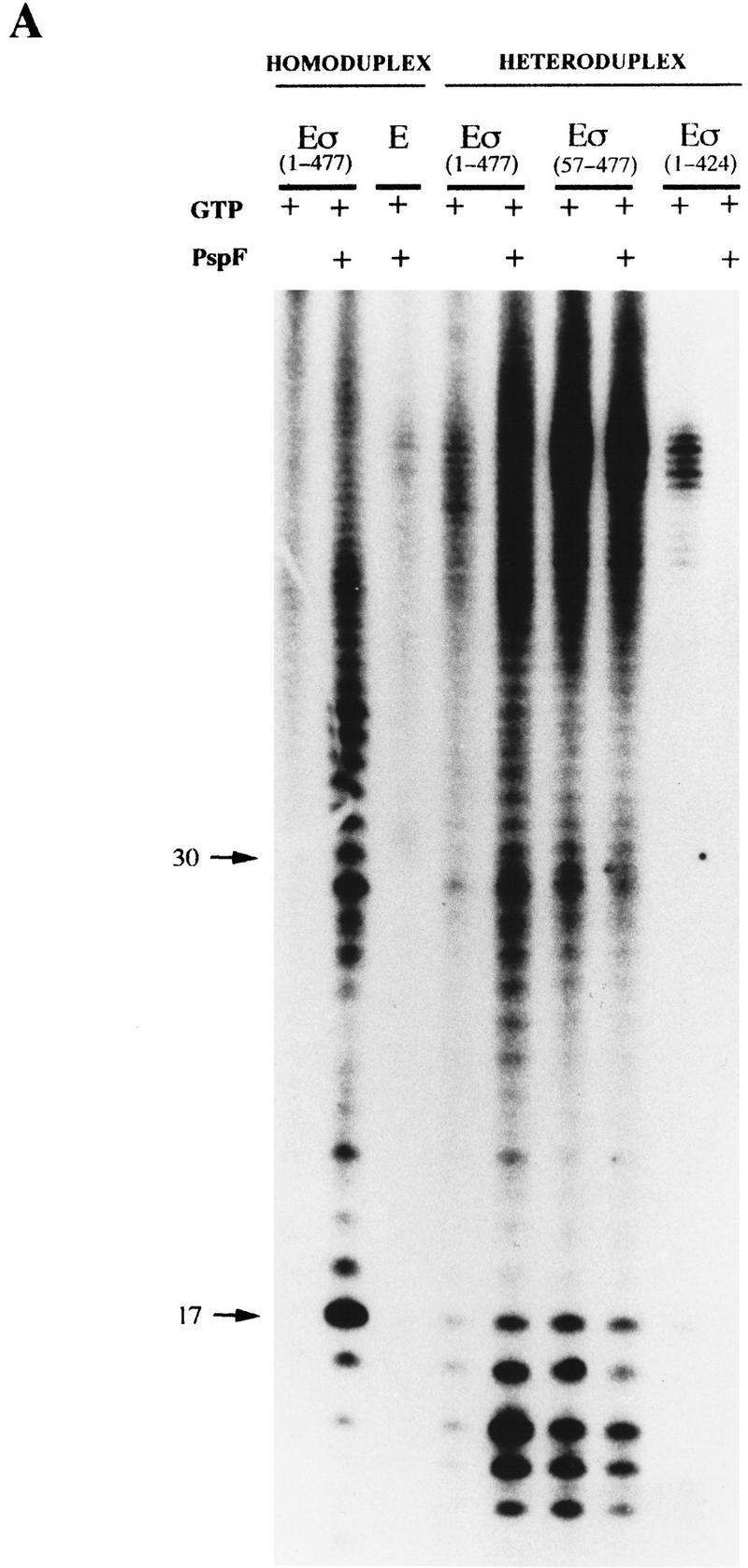
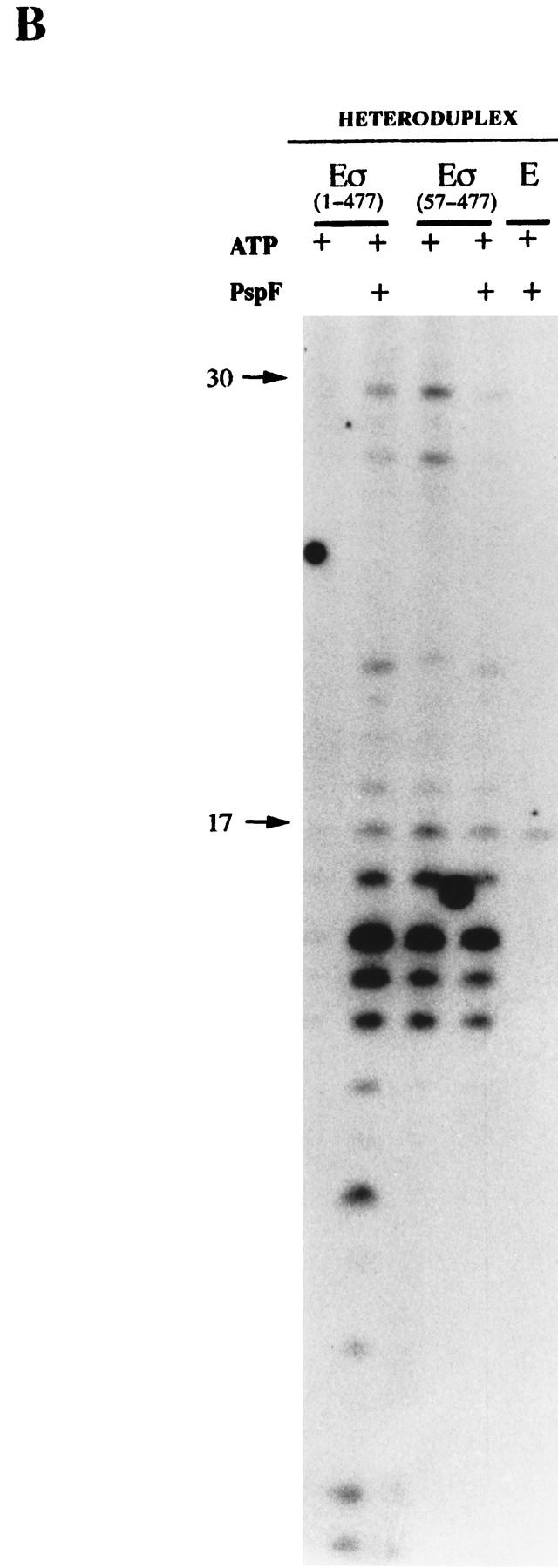
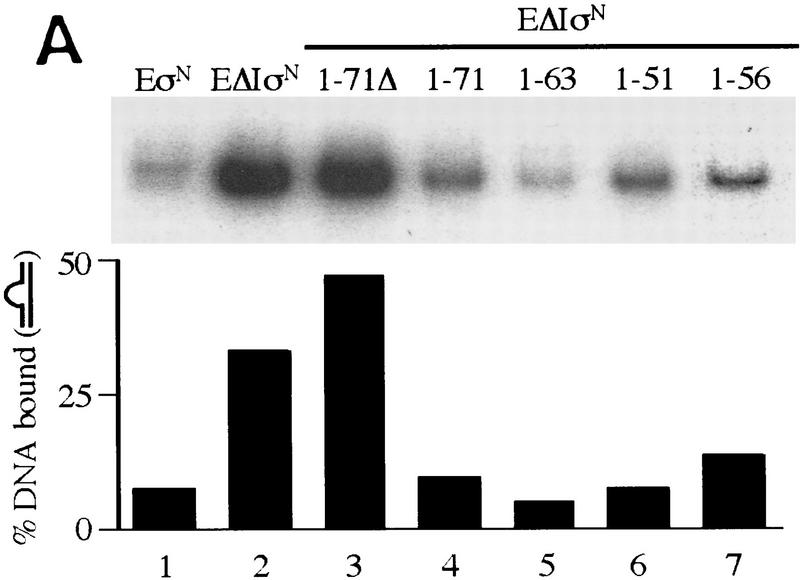
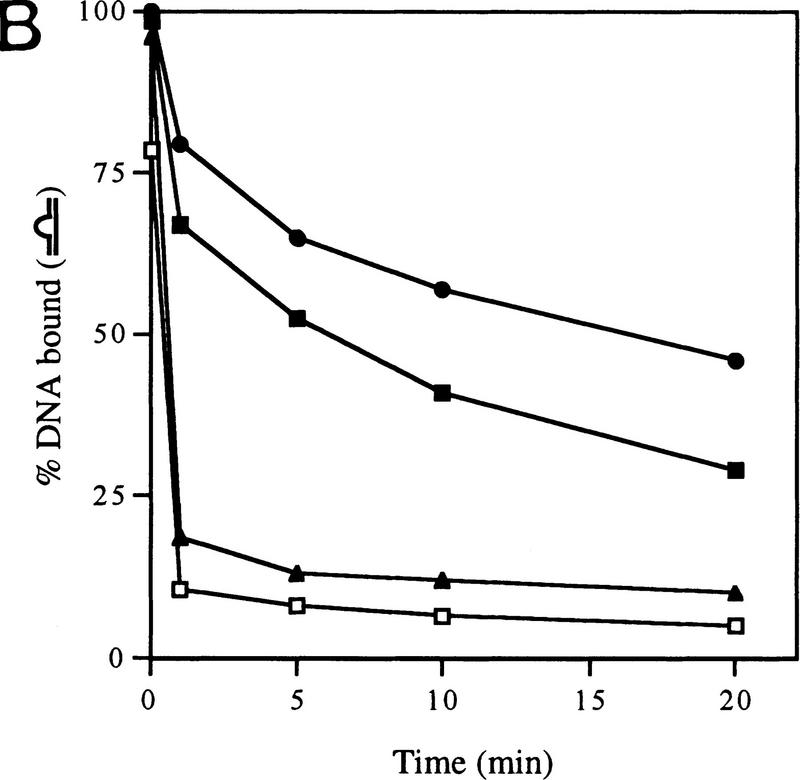
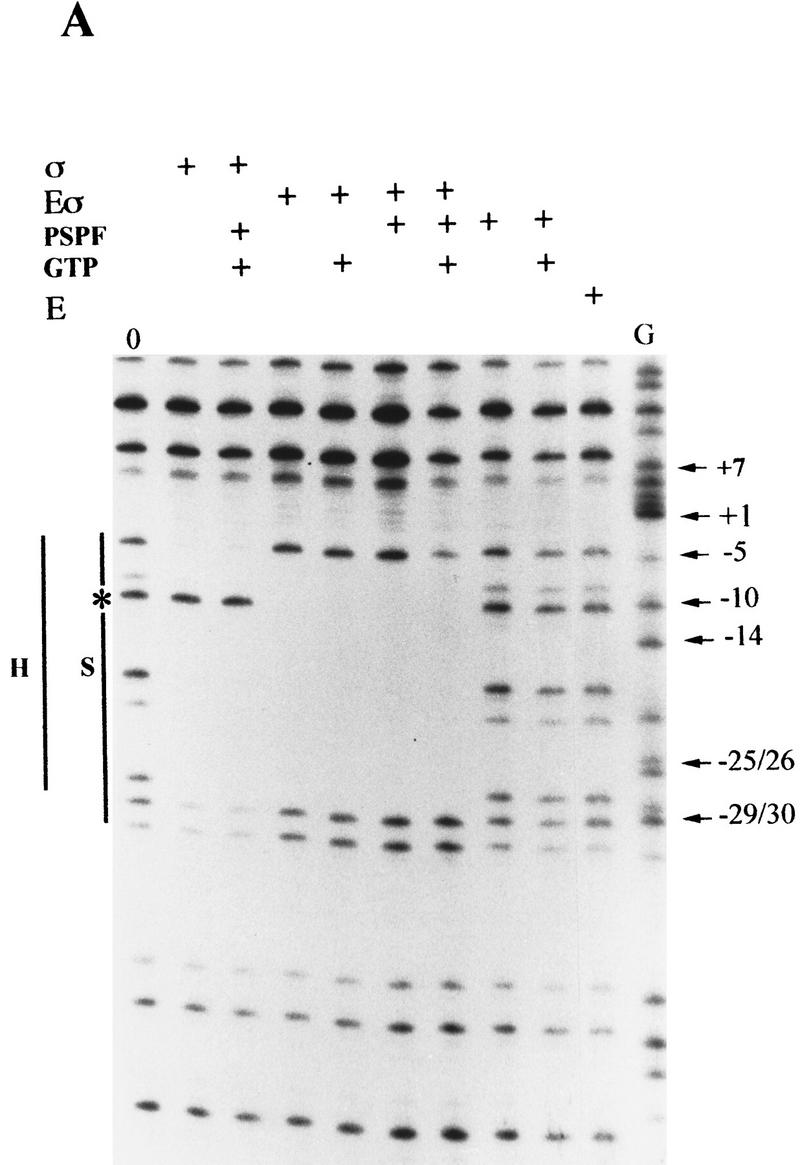
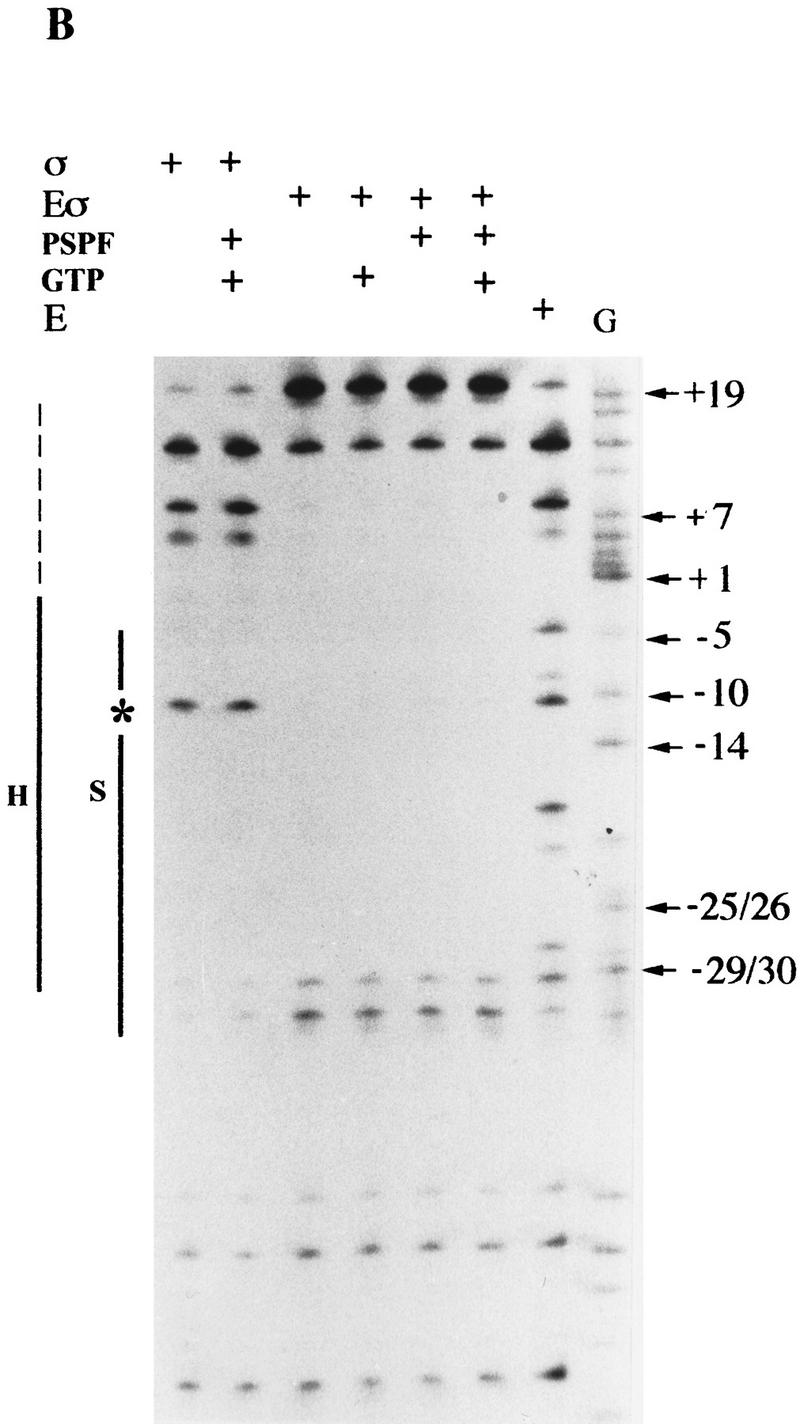
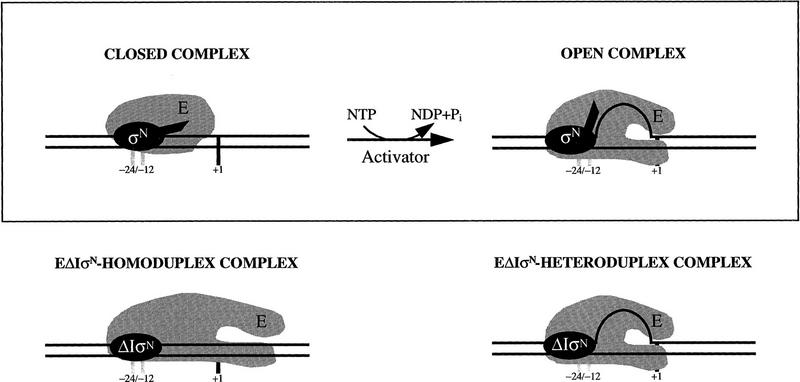
In bacteria, association of the specialized sigmaN protein with the core RNA polymerase subunits forms a holoenzyme able to bind promoter DNA, but unable to melt DNA and initiate transcription unless acted on by an activator protein. The conserved amino-terminal 50 amino acids of sigmaN (Region I) are required for the response to activators. We have used pre-melted DNA templates, in which the template strand is unpaired and accessible for transcription initiation, to mimic a naturally melted promoter and explore the function of Region I. Our results indicate that one activity of Region I sequences is to inhibit productive interaction of holoenzyme with pre-melted DNA. On pre-melted DNA targets, either activation of sigmaN-holoenzyme or removal of Region I allowed efficient formation of complexes in which melted DNA was sequestered by RNA polymerase. Like natural pre-initiation complexes formed on conventional DNA templates through the action of activator, such complexes were heparin-resistant and transcriptionally active. The inhibitory sigmaN Region I domain functioned in trans to confer heparin sensitivity to complexes between Region I-deleted holoenzyme and pre-melted promoter DNA. Evidence that Region I senses the conformation of the promoter was obtained from protein footprint experiments. We suggest that one function for Region I is to mask a single-strand DNA-binding activity of the holoenzyme. On the basis of extended DNA footprints of Region I-deleted holoenzyme, we also propose that Region I prevents RNA polymerase isomerization, a conformational change necessary for access to and the subsequent stable association of holoenzyme with melted DNA.
Figures
References
-
- Buck M, Cannon W. Specific binding of the transcription factor σ-54 to promoter DNA. Nature. 1992;358:422–424. - PubMed
-
- Burgess R, Travers A, Dunn J, Bautz E. Factor stimulating transcription by RNA polymerase. Nature. 1969;221:43–46. - PubMed
-
- Cannon W, Claverie-Martin F, Austin S, Buck M. Identification of a DNA-contacting surface in the transcription factor σ54 (σN) Mol Microbiol. 1994;11:227–236. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources