

Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents
- PMID: 32241904
- PMCID: PMC7677483
- DOI: 10.1136/gutjnl-2019-319766
Aberrant gut microbiota alters host metabolome and impacts renal failure in humans and rodents
Abstract
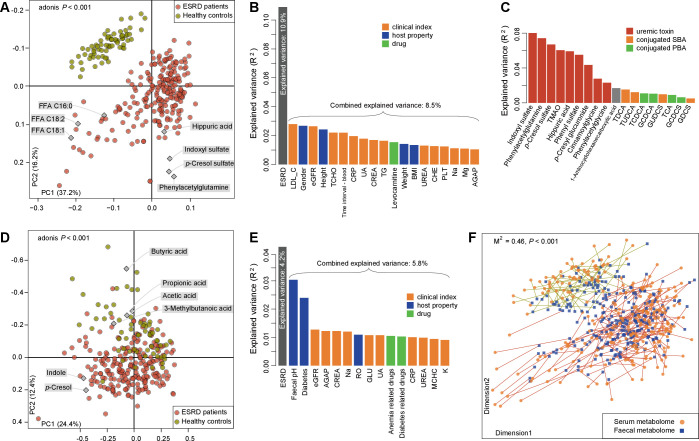
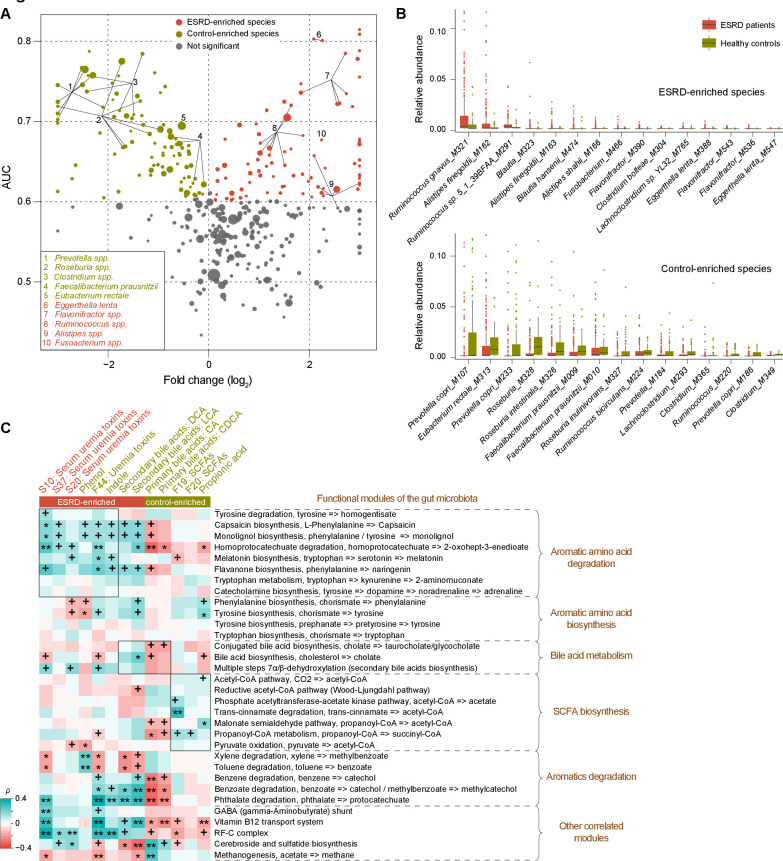
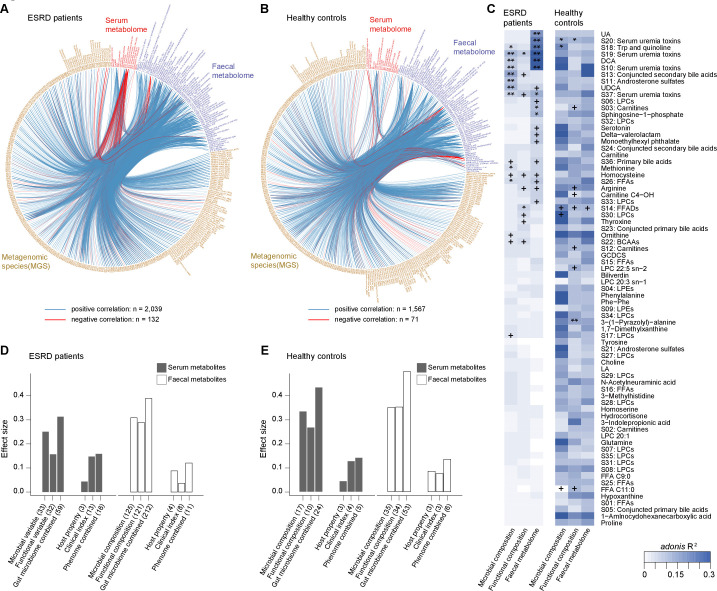
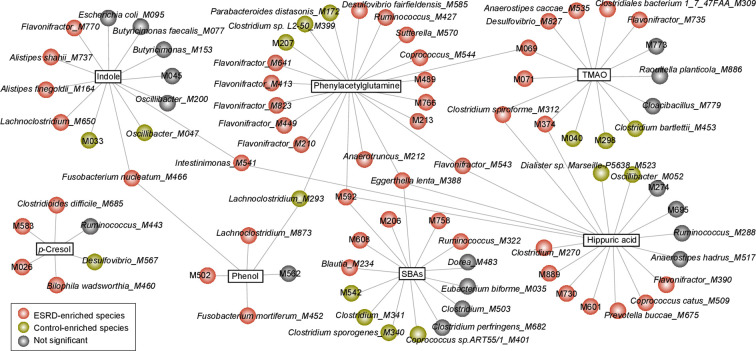
Objective: Patients with renal failure suffer from symptoms caused by uraemic toxins, possibly of gut microbial origin, as deduced from studies in animals. The aim of the study is to characterise relationships between the intestinal microbiome composition, uraemic toxins and renal failure symptoms in human end-stage renal disease (ESRD).
Design: Characterisation of gut microbiome, serum and faecal metabolome and human phenotypes in a cohort of 223 patients with ESRD and 69 healthy controls. Multidimensional data integration to reveal links between these datasets and the use of chronic kidney disease (CKD) rodent models to test the effects of intestinal microbiome on toxin accumulation and disease severity.
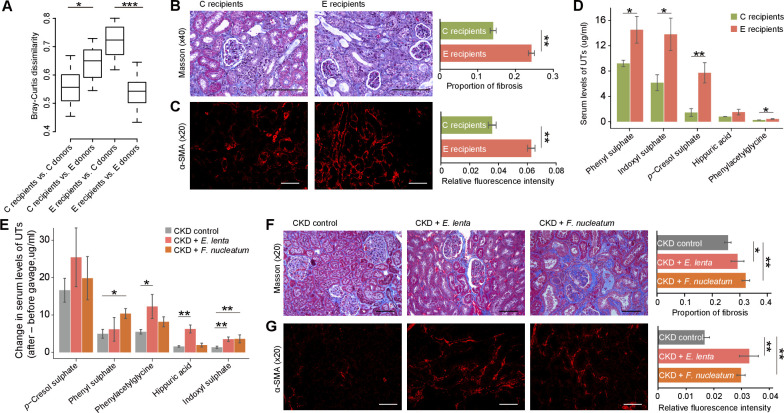
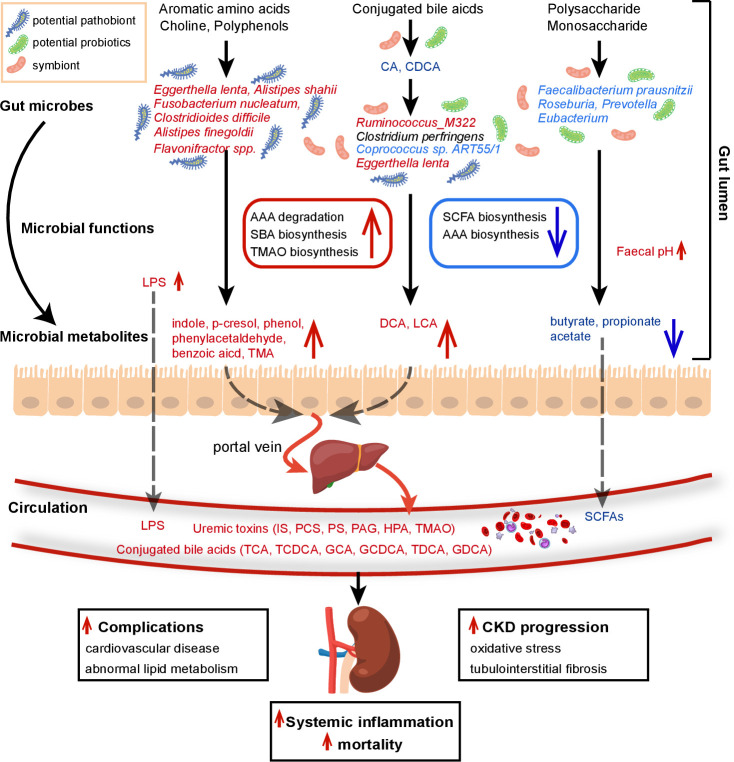
Results: A group of microbial species enriched in ESRD correlates tightly to patient clinical variables and encode functions involved in toxin and secondary bile acids synthesis; the relative abundance of the microbial functions correlates with the serum or faecal concentrations of these metabolites. Microbiota from patients transplanted to renal injured germ-free mice or antibiotic-treated rats induce higher production of serum uraemic toxins and aggravated renal fibrosis and oxidative stress more than microbiota from controls. Two of the species, Eggerthella lenta and Fusobacterium nucleatum, increase uraemic toxins production and promote renal disease development in a CKD rat model. A probiotic Bifidobacterium animalis decreases abundance of these species, reduces levels of toxins and the severity of the disease in rats.
Conclusion: Aberrant gut microbiota in patients with ESRD sculpts a detrimental metabolome aggravating clinical outcomes, suggesting that the gut microbiota will be a promising target for diminishing uraemic toxicity in those patients.
Trial registration number: This study was registered at ClinicalTrials.gov (NCT03010696).
Keywords: bile acid; enteric bacterial microflora.
© Author(s) (or their employer(s)) 2020. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
Similar articles
-
Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach.Kidney Int. 2017 Sep;92(3):634-645. doi: 10.1016/j.kint.2017.02.011. Epub 2017 Apr 8. Kidney Int. 2017. PMID: 28396122
-
Longitudinal analysis of fecal microbiome and metabolome during renal fibrotic progression in a unilateral ureteral obstruction animal model.Eur J Pharmacol. 2020 Nov 5;886:173555. doi: 10.1016/j.ejphar.2020.173555. Epub 2020 Sep 14. Eur J Pharmacol. 2020. PMID: 32937112
-
Effects of Fecal Microbiota Transplantation on Composition in Mice with CKD.Toxins (Basel). 2020 Nov 24;12(12):741. doi: 10.3390/toxins12120741. Toxins (Basel). 2020. PMID: 33255454 Free PMC article.
-
Oral iron supplementation: Potential implications for the gut microbiome and metabolome in patients with CKD.Hemodial Int. 2017 Jun;21 Suppl 1:S28-S36. doi: 10.1111/hdi.12553. Epub 2017 Mar 22. Hemodial Int. 2017. PMID: 28328083 Review.
-
Gut microbiota in chronic kidney disease.Nefrologia. 2017 Jan-Feb;37(1):9-19. doi: 10.1016/j.nefro.2016.05.008. Epub 2016 Aug 21. Nefrologia. 2017. PMID: 27553986 Review. English, Spanish.
Cited by
-
Circulating concentrations of bile acids and prevalent chronic kidney disease among newly diagnosed type 2 diabetes: a cross-sectional study.Nutr J. 2024 Mar 2;23(1):28. doi: 10.1186/s12937-024-00928-2. Nutr J. 2024. PMID: 38429722 Free PMC article.
-
Natural products from plants and microorganisms: Novel therapeutics for chronic kidney disease via gut microbiota regulation.Front Pharmacol. 2023 Jan 17;13:1068613. doi: 10.3389/fphar.2022.1068613. eCollection 2022. Front Pharmacol. 2023. PMID: 36733377 Free PMC article. Review.
-
Causal Effects of Specific Gut Microbiota on Chronic Kidney Diseases and Renal Function-A Two-Sample Mendelian Randomization Study.Nutrients. 2023 Jan 11;15(2):360. doi: 10.3390/nu15020360. Nutrients. 2023. PMID: 36678231 Free PMC article.
-
Therapeutic Potential of Photobiomodulation for Chronic Kidney Disease.Int J Mol Sci. 2022 Jul 21;23(14):8043. doi: 10.3390/ijms23148043. Int J Mol Sci. 2022. PMID: 35887386 Free PMC article. Review.
-
Gut Microbiota as a Source of Uremic Toxins.Int J Mol Sci. 2022 Jan 1;23(1):483. doi: 10.3390/ijms23010483. Int J Mol Sci. 2022. PMID: 35008909 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases