

Virome diversity shaped by genetic evolution and ecological landscape of Haemaphysalis longicornis
- PMID: 38378577
- PMCID: PMC10880243
- DOI: 10.1186/s40168-024-01753-9
Virome diversity shaped by genetic evolution and ecological landscape of Haemaphysalis longicornis
Abstract
Background: Haemaphysalis longicornis is drawing attentions for its geographic invasion, extending population, and emerging disease threat. However, there are still substantial gaps in our knowledge of viral composition in relation to genetic diversity of H. longicornis and ecological factors, which are important for us to understand interactions between virus and vector, as well as between vector and ecological elements.
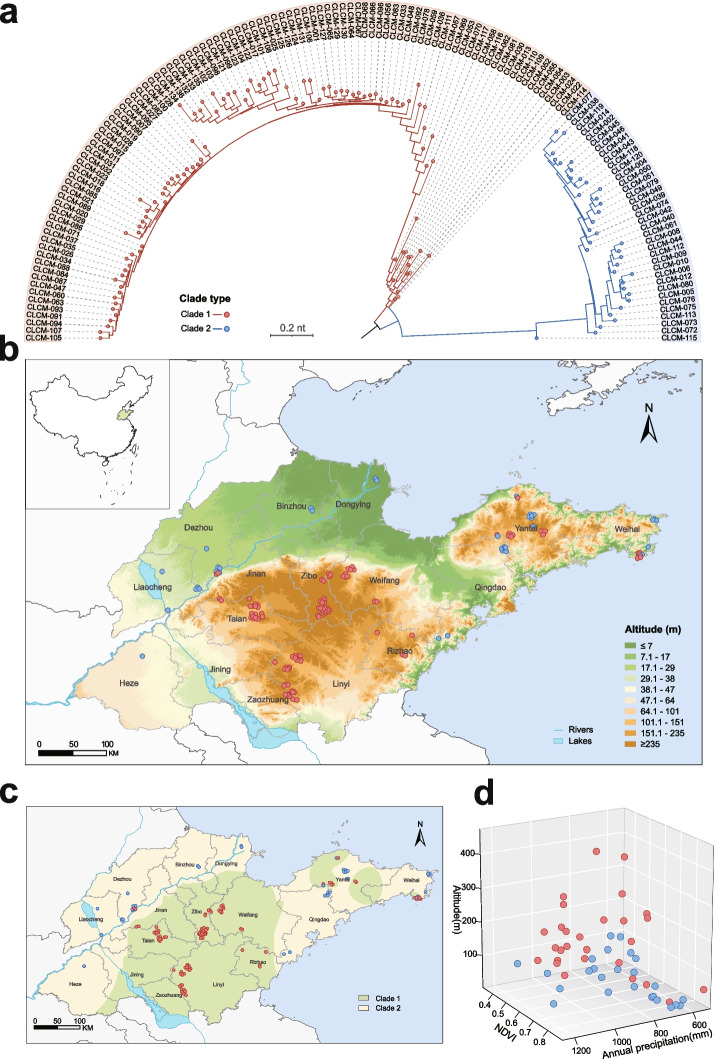
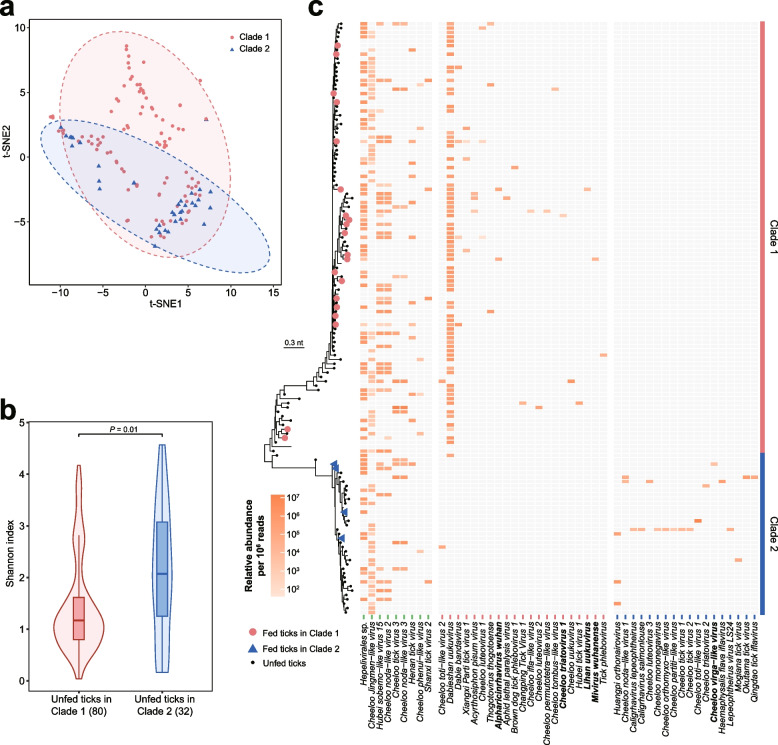
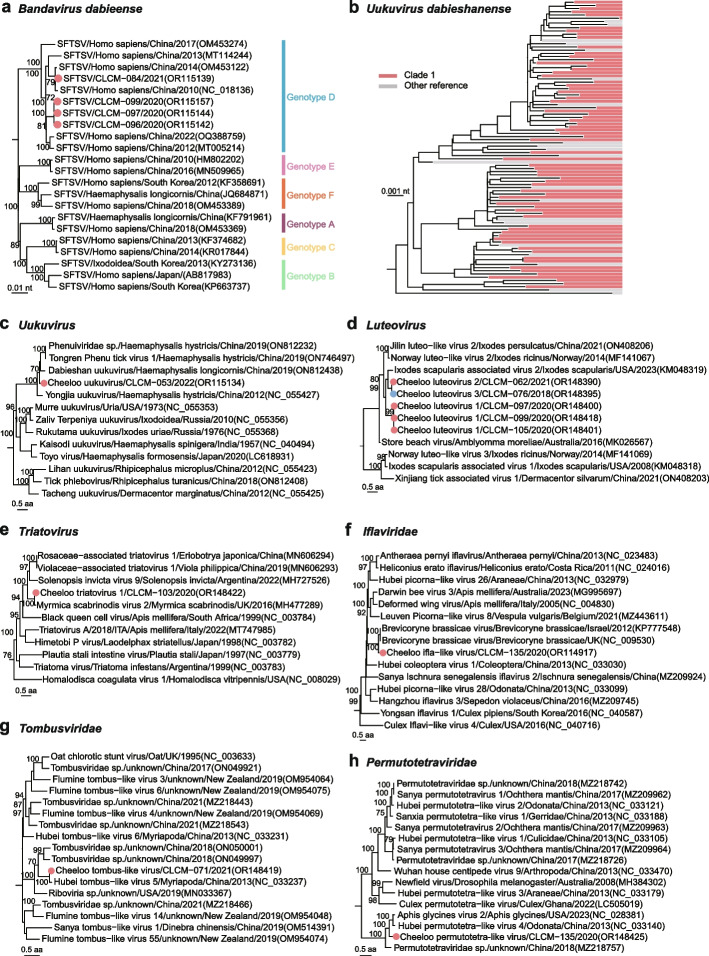
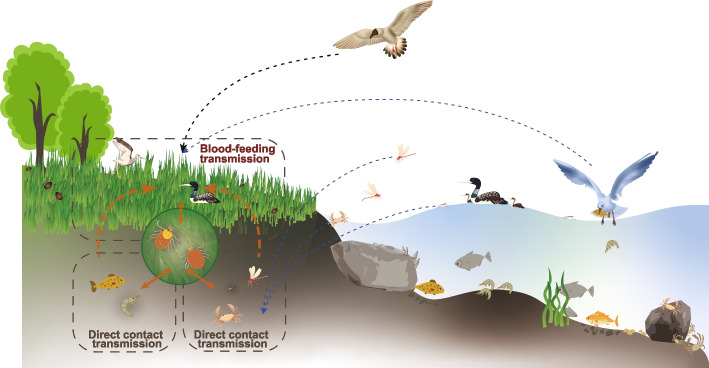
Results: We conducted the meta-transcriptomic sequencing of 136 pools of H. longicornis and identified 508 RNA viruses of 48 viral species, 22 of which have never been reported. Phylogenetic analysis of mitochondrion sequences divided the ticks into two genetic clades, each of which was geographically clustered and significantly associated with ecological factors, including altitude, precipitation, and normalized difference vegetation index. The two clades showed significant difference in virome diversity and shared about one fifth number of viral species that might have evolved to "generalists." Notably, Bandavirus dabieense, the pathogen of severe fever with thrombocytopenia syndrome was only detected in ticks of clade 1, and half number of clade 2-specific viruses were aquatic-animal-associated.
Conclusions: These findings highlight that the virome diversity is shaped by internal genetic evolution and external ecological landscape of H. longicornis and provide the new foundation for promoting the studies on virus-vector-ecology interaction and eventually for evaluating the risk of H. longicornis for transmitting the viruses to humans and animals. Video Abstract.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
Viromes of Haemaphysalis longicornis reveal different viral abundance and diversity in free and engorged ticks.Virol Sin. 2024 Apr;39(2):194-204. doi: 10.1016/j.virs.2024.02.003. Epub 2024 Feb 13. Virol Sin. 2024. PMID: 38360150 Free PMC article.
-
Isolation of Severe Fever with Thrombocytopenia Syndrome Virus from Various Tick Species in Area with Human Severe Fever with Thrombocytopenia Syndrome Cases.Vector Borne Zoonotic Dis. 2021 May;21(5):378-384. doi: 10.1089/vbz.2020.2720. Epub 2021 Feb 3. Vector Borne Zoonotic Dis. 2021. PMID: 33535015
-
First Isolation of Severe Fever with Thrombocytopenia Syndrome Virus from Haemaphysalis longicornis Ticks Collected in Severe Fever with Thrombocytopenia Syndrome Outbreak Areas in the Republic of Korea.Vector Borne Zoonotic Dis. 2016 Jan;16(1):66-70. doi: 10.1089/vbz.2015.1832. Epub 2016 Jan 8. Vector Borne Zoonotic Dis. 2016. PMID: 26745758 Free PMC article.
-
Haemaphysalis longicornis Ticks as Reservoir and Vector of Severe Fever with Thrombocytopenia Syndrome Virus in China.Emerg Infect Dis. 2015 Oct;21(10):1770-6. doi: 10.3201/eid2110.150126. Emerg Infect Dis. 2015. PMID: 26402039 Free PMC article.
-
The Ecology of New Constituents of the Tick Virome and Their Relevance to Public Health.Viruses. 2019 Jun 7;11(6):529. doi: 10.3390/v11060529. Viruses. 2019. PMID: 31181599 Free PMC article. Review.
Cited by
-
Virome specific to tick genus with distinct ecogeographical distribution.Microbiome. 2025 Feb 28;13(1):57. doi: 10.1186/s40168-025-02061-6. Microbiome. 2025. PMID: 40022268 Free PMC article.
-
Alarming implications: severe fever with thrombocytopenia syndrome and its biological vectors in the context of climate change.Front Microbiol. 2025 Jul 25;16:1544427. doi: 10.3389/fmicb.2025.1544427. eCollection 2025. Front Microbiol. 2025. PMID: 40785782 Free PMC article. Review.
-
Sexual epigenetics: genome-wide analysis revealed differential DNA methylation in the vector tick Haemaphysalis longicornis.Parasit Vectors. 2025 Jun 1;18(1):202. doi: 10.1186/s13071-025-06810-2. Parasit Vectors. 2025. PMID: 40452039 Free PMC article.
-
RNA viromes of Dermacentor nuttalli ticks reveal a novel uukuvirus in Qīnghăi Province, China.Virol Sin. 2024 Aug;39(4):537-545. doi: 10.1016/j.virs.2024.04.006. Epub 2024 Apr 26. Virol Sin. 2024. PMID: 38679334 Free PMC article.
-
Host taxonomy and environment shapes insectivore viromes and viral spillover risks in Southwestern China.Microbiome. 2025 May 16;13(1):122. doi: 10.1186/s40168-025-02115-9. Microbiome. 2025. PMID: 40380277 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
