

Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation
- PMID: 23541343
- PMCID: PMC3617388
- DOI: 10.1016/j.ajhg.2013.03.004
Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation
Abstract
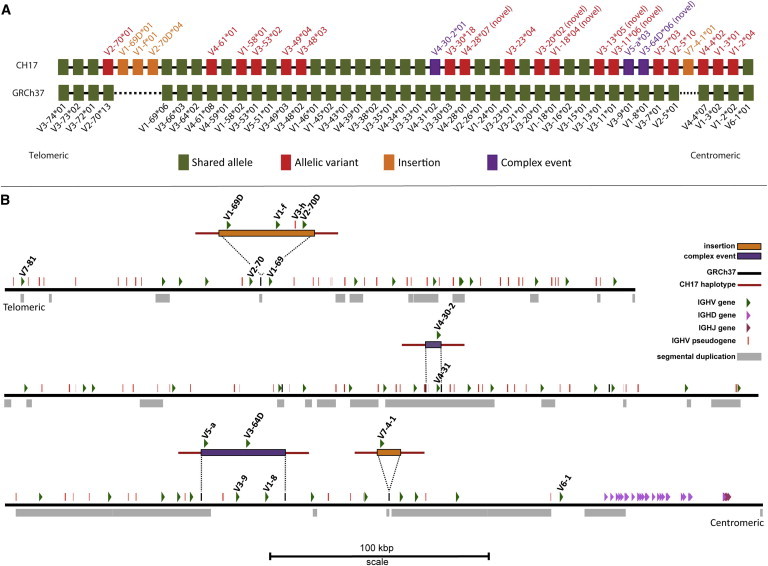
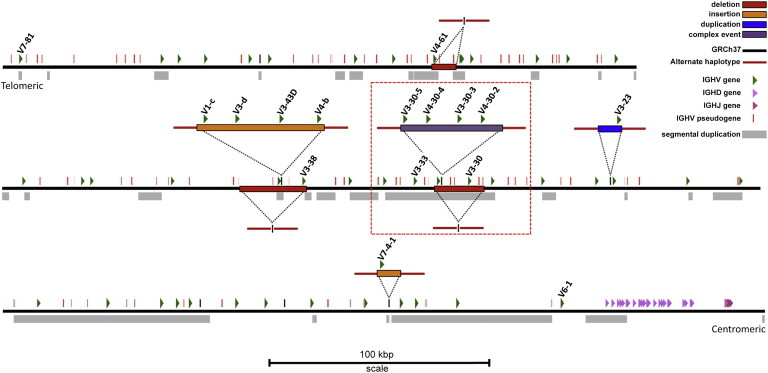
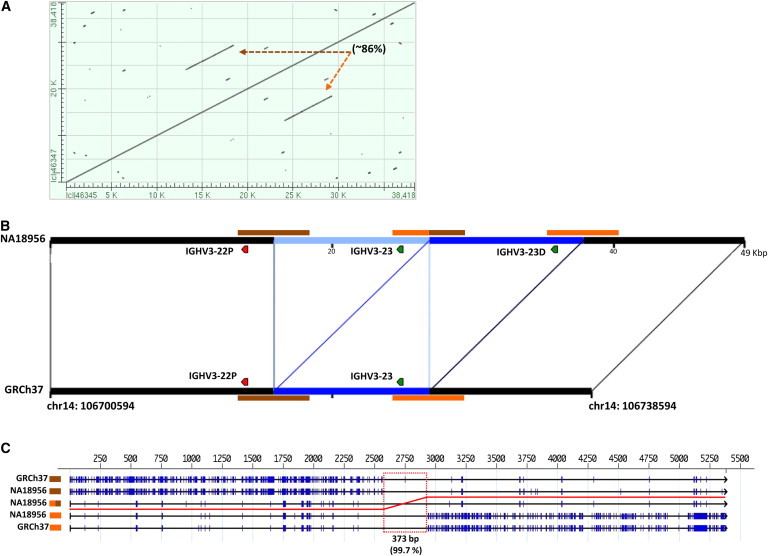
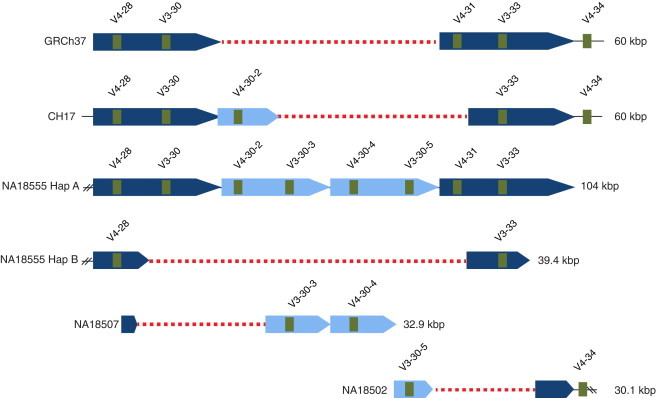
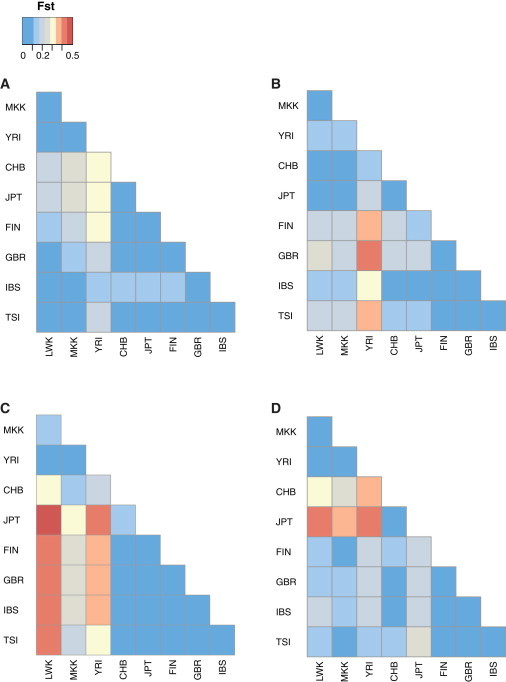
The immunoglobulin heavy-chain locus (IGH) encodes variable (IGHV), diversity (IGHD), joining (IGHJ), and constant (IGHC) genes and is responsible for antibody heavy-chain biosynthesis, which is vital to the adaptive immune response. Programmed V-(D)-J somatic rearrangement and the complex duplicated nature of the locus have impeded attempts to reconcile its genomic organization based on traditional B-lymphocyte derived genetic material. As a result, sequence descriptions of germline variation within IGHV are lacking, haplotype inference using traditional linkage disequilibrium methods has been difficult, and the human genome reference assembly is missing several expressed IGHV genes. By using a hydatidiform mole BAC clone resource, we present the most complete haplotype of IGHV, IGHD, and IGHJ gene regions derived from a single chromosome, representing an alternate assembly of ∼1 Mbp of high-quality finished sequence. From this we add 101 kbp of previously uncharacterized sequence, including functional IGHV genes, and characterize four large germline copy-number variants (CNVs). In addition to this germline reference, we identify and characterize eight CNV-containing haplotypes from a panel of nine diploid genomes of diverse ethnic origin, discovering previously unmapped IGHV genes and an additional 121 kbp of insertion sequence. We genotype four of these CNVs by using PCR in 425 individuals from nine human populations. We find that all four are highly polymorphic and show considerable evidence of stratification (Fst = 0.3-0.5), with the greatest differences observed between African and Asian populations. These CNVs exhibit weak linkage disequilibrium with SNPs from two commercial arrays in most of the populations tested.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Tuzun E., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
