

Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut
- PMID: 10543789
- PMCID: PMC91647
- DOI: 10.1128/AEM.65.11.4799-4807.1999
Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut
Abstract
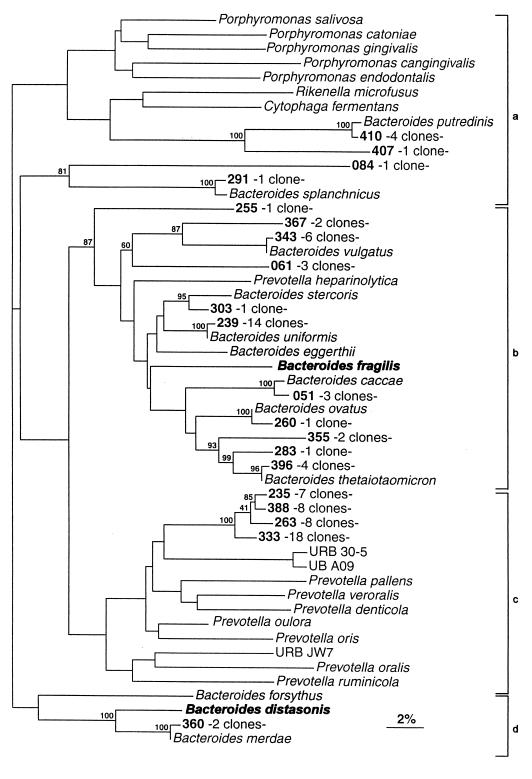
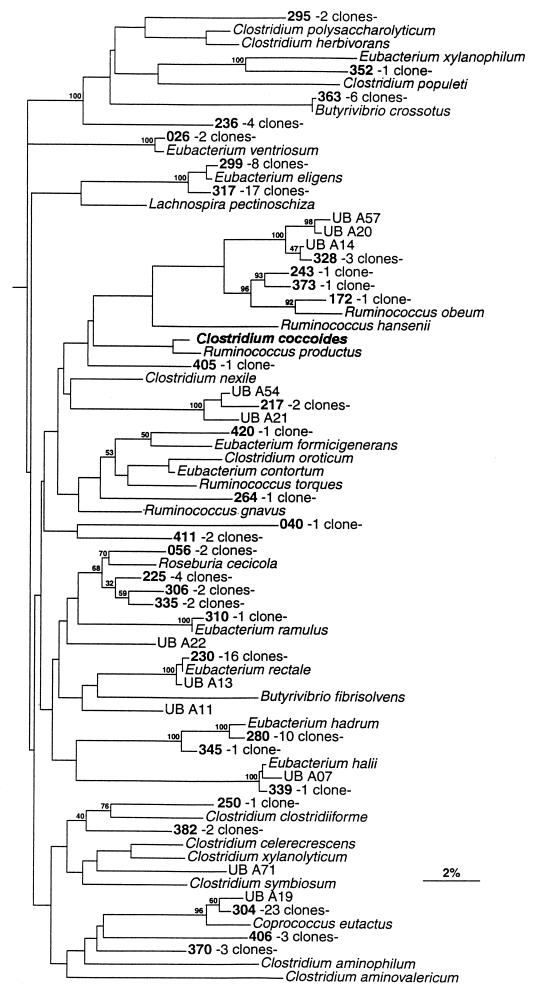
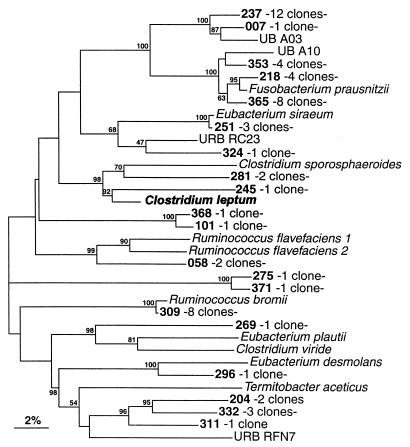
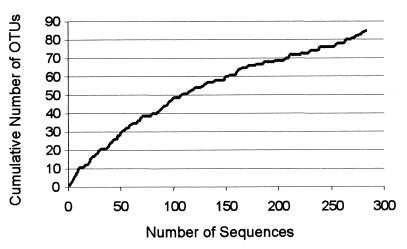
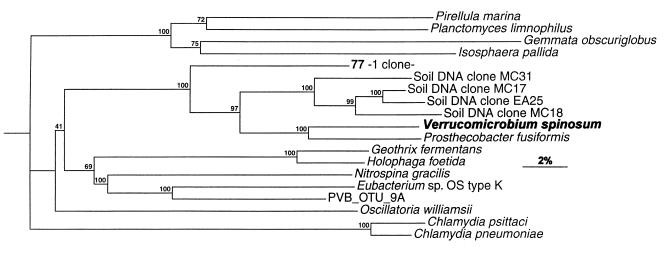
The human intestinal tract harbors a complex microbial ecosystem which plays a key role in nutrition and health. Although this microbiota has been studied in great detail by culture techniques, microscopic counts on human feces suggest that 60 to 80% of the observable bacteria cannot be cultivated. Using comparative analysis of cloned 16S rRNA gene (rDNA) sequences, we have investigated the bacterial diversity (both cultivated and noncultivated bacteria) within an adult-male fecal sample. The 284 clones obtained from 10-cycle PCR were classified into 82 molecular species (at least 98% similarity). Three phylogenetic groups contained 95% of the clones: the Bacteroides group, the Clostridium coccoides group, and the Clostridium leptum subgroup. The remaining clones were distributed among a variety of phylogenetic clusters. Only 24% of the molecular species recovered corresponded to described organisms (those whose sequences were available in public databases), and all of these were established members of the dominant human fecal flora (e.g., Bacteroides thetaiotaomicron, Fusobacterium prausnitzii, and Eubacterium rectale). However, the majority of generated rDNA sequences (76%) did not correspond to known organisms and clearly derived from hitherto unknown species within this human gut microflora.
Figures
References
-
- Collins M D, Lawson P A, Willems A, Cordoba J J, Fernandez-Garayzabal J, Garcia P, Cai J, Hippe H, Farrow J A. The phylogeny of the genus Clostridium: proposal of five new general and eleven new species combinations. Int J Syst Bacteriol. 1994;44:812–826. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
