

Comparative genomics of foot-and-mouth disease virus
- PMID: 15858032
- PMCID: PMC1091679
- DOI: 10.1128/JVI.79.10.6487-6504.2005
Comparative genomics of foot-and-mouth disease virus
Abstract
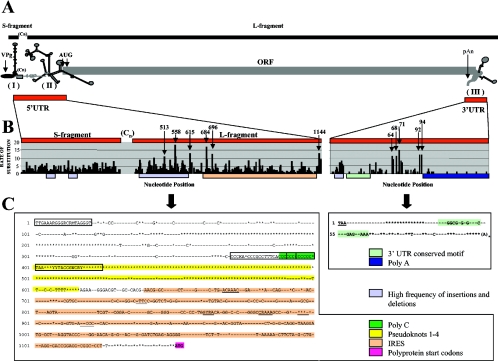
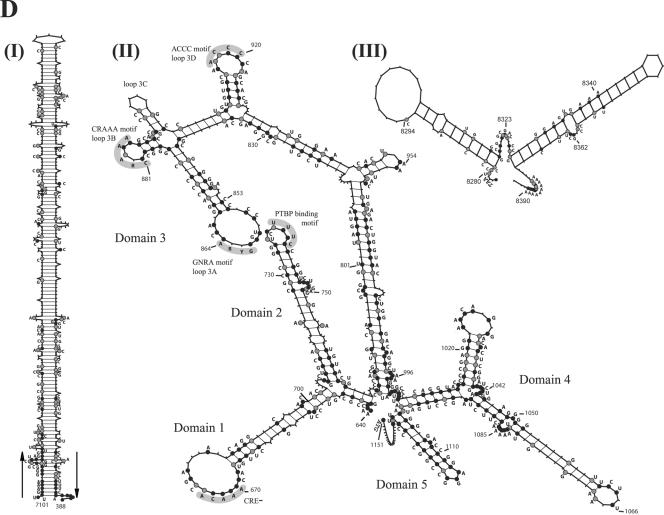
Here we present complete genome sequences, including a comparative analysis, of 103 isolates of foot-and-mouth disease virus (FMDV) representing all seven serotypes and including the first complete sequences of the SAT1 and SAT3 genomes. The data reveal novel highly conserved genomic regions, indicating functional constraints for variability as well as novel viral genomic motifs with likely biological relevance. Previously undescribed invariant motifs were identified in the 5' and 3' untranslated regions (UTR), as was tolerance for insertions/deletions in the 5' UTR. Fifty-eight percent of the amino acids encoded by FMDV isolates are invariant, suggesting that these residues are critical for virus biology. Novel, conserved sequence motifs with likely functional significance were identified within proteins L(pro), 1B, 1D, and 3C. An analysis of the complete FMDV genomes indicated phylogenetic incongruities between different genomic regions which were suggestive of interserotypic recombination. Additionally, a novel SAT virus lineage containing nonstructural protein-encoding regions distinct from other SAT and Euroasiatic lineages was identified. Insights into viral RNA sequence conservation and variability and genetic diversity in nature will likely impact our understanding of FMDV infections, host range, and transmission.
Figures

References
-
- Acharya, R., E. Fry, D. Stuart, G. Fox, D. Rowlands, and F. Brown. 1990. The structure of foot-and-mouth disease virus: implications for its physical and biological properties. Vet. Microbiol. 23:21-34. - PubMed
-
- Acharya, R., E. Fry, D. Stuart, G. Fox, D. Rowlands, and F. Brown. 1989. The three-dimensional structure of foot-and-mouth disease virus at 2.9 Å resolution. Nature (London) 337:709-716. - PubMed
-
- Agol, V. I., A. V. Paul, and E. Wimmer. 1999. Paradoxes of the replication of picornaviral genomes. Virus Res. 62:129-147. - PubMed
-
- Ambros, V., and D. Baltimore. 1978. Protein is linked to the 5′ end of poliovirus RNA by a phosphodiester linkage to tyrosine. J. Biol. Chem. 253:5263-5266. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
