

Novel kingdom-level eukaryotic diversity in anoxic environments
- PMID: 12060775
- PMCID: PMC123066
- DOI: 10.1073/pnas.062169599
Novel kingdom-level eukaryotic diversity in anoxic environments
Abstract
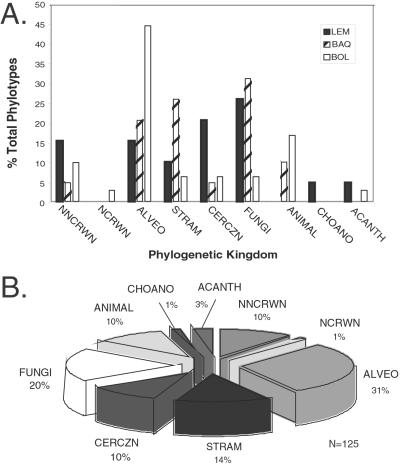
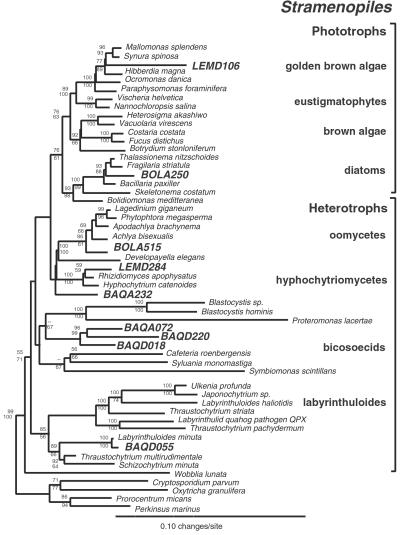
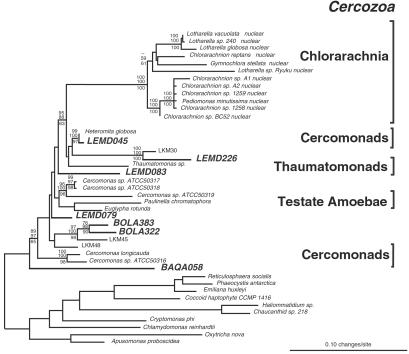
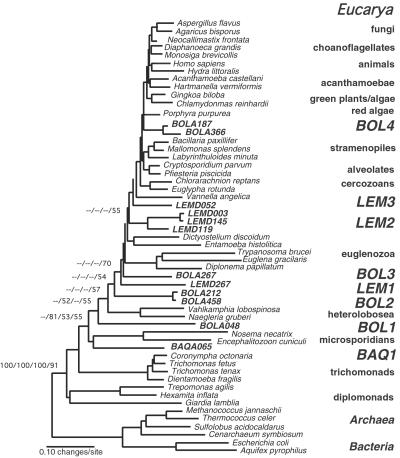
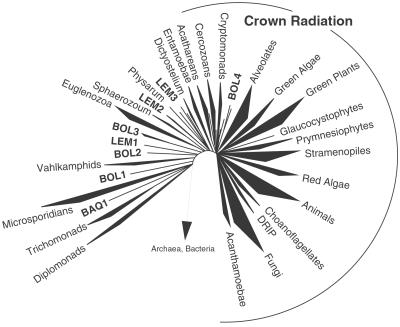
Molecular evolutionary studies of eukaryotes have relied on a sparse collection of gene sequences that do not represent the full range of eukaryotic diversity in nature. Anaerobic microbes, particularly, have had little representation in phylogenetic studies. Such organisms are the least known of eukaryotes and probably are the most phylogenetically diverse. To provide fresh perspective on the natural diversity of eukaryotes in anoxic environments and also to discover novel sequences for evolutionary studies, we conducted a cultivation-independent, molecular phylogenetic survey of three anoxic sediments, including both freshwater and marine samples. Many previously unrecognized eukaryotes were identified, including representatives of seven lineages that are not specifically related to any known organisms at the kingdom-level and branch below the eukaryotic "crown" radiation of animals, plants, fungi, stramenopiles, etc. The survey additionally identified new sequences characteristic of known ecologically important eukaryotic groups with anaerobic members. Phylogenetic analyses with the new sequences enhance our understanding of the diversity and pattern of eukaryotic evolution.
Figures
References
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
