

Human immunodeficiency virus type 1 env sequences from Calcutta in eastern India: identification of features that distinguish subtype C sequences in India from other subtype C sequences
- PMID: 11581417
- PMCID: PMC114623
- DOI: 10.1128/JVI.75.21.10479-10487.2001
Human immunodeficiency virus type 1 env sequences from Calcutta in eastern India: identification of features that distinguish subtype C sequences in India from other subtype C sequences
Abstract
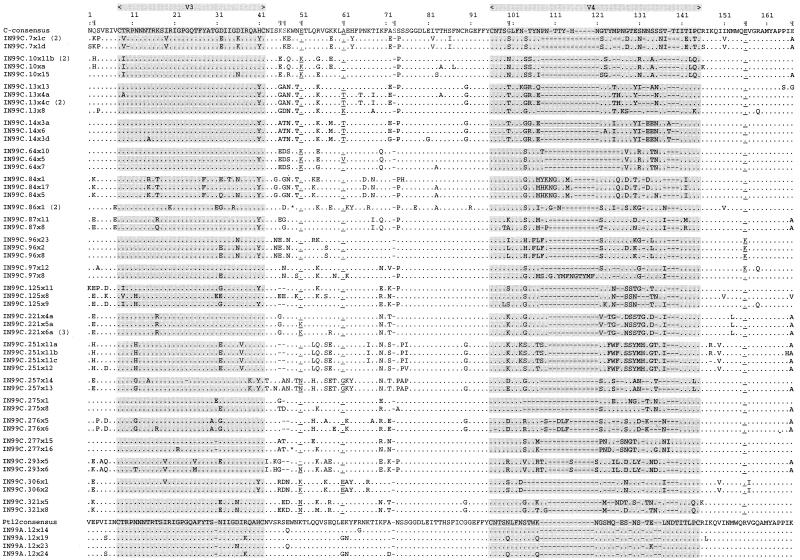
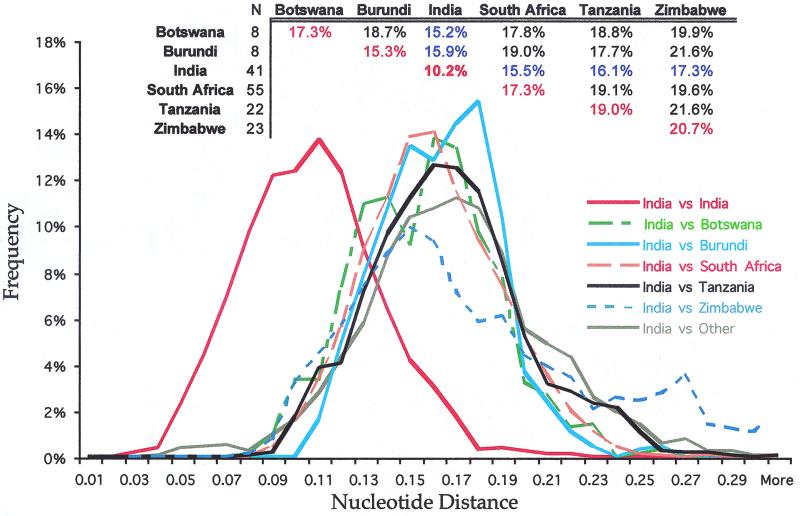
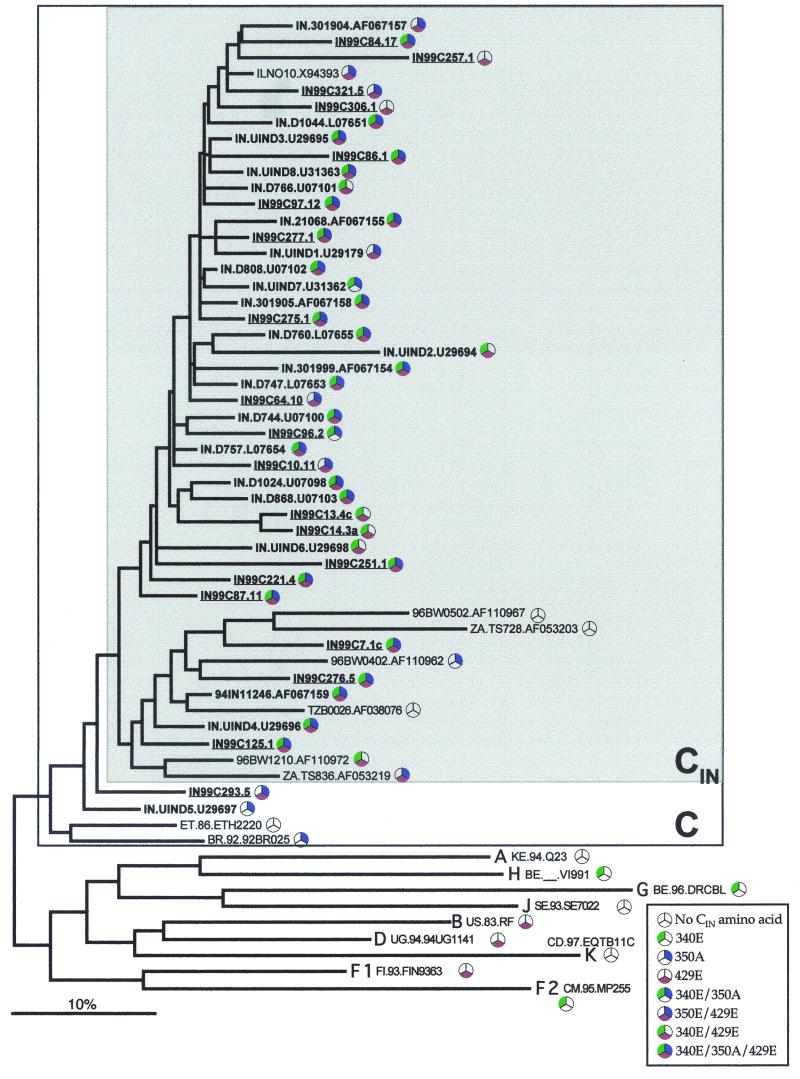
India is experiencing a rapid spread of human immunodeficiency virus type 1 (HIV-1), primarily through heterosexual transmission of subtype C viruses. To delineate the molecular features of HIV-1 circulating in India, we sequenced the V3-V4 region of viral env from 21 individuals attending an HIV clinic in Calcutta, the most populous city in the eastern part of the country, and analyzed these and the other Indian sequences in the HIV database. Twenty individuals were infected with viruses having a subtype C env, and one had viruses with a subtype A env. Analyses of 192 subtype C sequences that included one sequence for each subject from this study and from the HIV database revealed that almost all sequences from India, along with a small number from other countries, form a phylogenetically distinct lineage within subtype C, which we designate C(IN). Overall, C(IN) lineage sequences were more closely related to each other (level of diversity, 10.2%) than to subtype C sequences from Botswana, Burundi, South Africa, Tanzania, and Zimbabwe (range, 15.3 to 20.7%). Of the three positions identified as signature amino acid substitution sites for C(IN) sequences (K340E, K350A, and G429E), 56% of the C(IN) sequences contained all three amino acids while 87% of the sequences contained at least two of these substitutions. Among the non-C(IN) sequences, all three amino acids were present in 2%, while 22% contained two or more of these amino acids. These results suggest that much of the current Indian epidemic is descended from a single introduction into the country. Identification of conserved signature amino acid positions could assist epidemiologic tracking and has implications for the development of a vaccine against subtype C HIV-1 in India.
Figures

References
-
- Abebe A, Pollakis G, Fontanet A L, Fisseha B, Tegbaru B, Kliphuis A, Tesfaye G, Negassa H, Cornelissen M, Goudsmit J, Renke de Wit T F. Identification of a genetic subcluster of HIV type 1 subtype C (C′) widespread in Ethiopia. AIDS Res Hum Retrovir. 2000;16:1909–1914. - PubMed
-
- Akaike H. A new look at statistical model identification. IEEE Trans Automatic Control. 1974;19:716–723.
-
- Baskar P V, Ray S C, Rao R, Quinn T C, Hildreth J E, Bollinger R C. Presence in India of HIV type 1 similar to North American strains. AIDS Res Hum Retrovir. 1994;10:1039–1041. - PubMed
-
- Beerli P, Grassly N C, Kuhner M K, Nickle D, Pybus O, Rain M, Rambaut A, Rodrigo A G, Wang Y. Population genetics of HIV: parameter estimation using genealogy-based methods. In: Rodrigo A G, Learn G H, editors. Computational and evolutionary analyses of HIV sequences. Boston, Mass: Kluwer Academic Publishers; 2001. pp. 217–258.
-
- Berger E, Doms R, Fenyo E, Korber B, Littman D, Moore J, Sattentau Q, Schuitemaker H, Sodroski J, Weiss R. A new classification for HIV-1. Nature. 1998;391:240. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
