

A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis
- PMID: 15372378
- PMCID: PMC1182111
- DOI: 10.1086/425287
A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis
Abstract
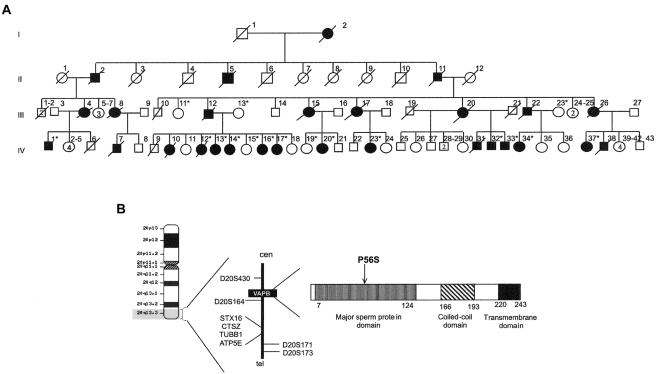
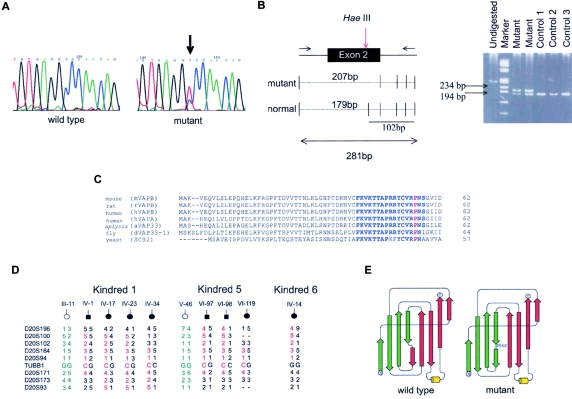
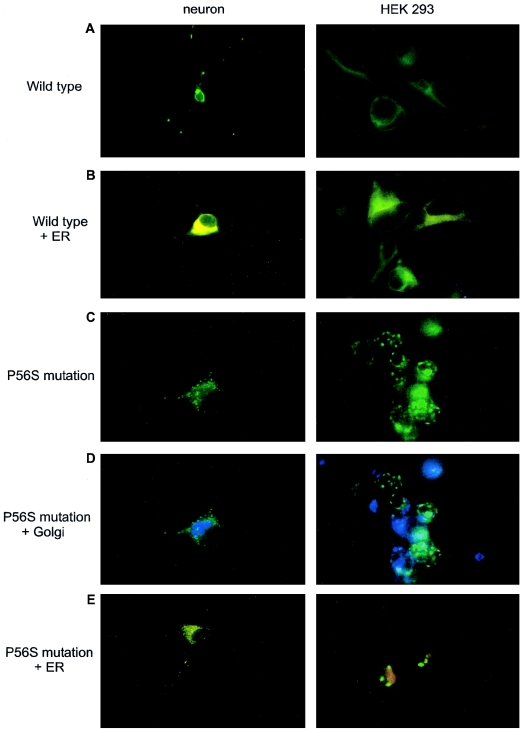
Motor neuron diseases (MNDs) are a group of neurodegenerative disorders with involvement of upper and/or lower motor neurons, such as amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), progressive bulbar palsy, and primary lateral sclerosis. Recently, we have mapped a new locus for an atypical form of ALS/MND (atypical amyotrophic lateral sclerosis [ALS8]) at 20q13.3 in a large white Brazilian family. Here, we report the finding of a novel missense mutation in the vesicle-associated membrane protein/synaptobrevin-associated membrane protein B (VAPB) gene in patients from this family. Subsequently, the same mutation was identified in patients from six additional kindreds but with different clinical courses, such as ALS8, late-onset SMA, and typical severe ALS with rapid progression. Although it was not possible to link all these families, haplotype analysis suggests a founder effect. Members of the vesicle-associated proteins are intracellular membrane proteins that can associate with microtubules and that have been shown to have a function in membrane transport. These data suggest that clinically variable MNDs may be caused by a dysfunction in intracellular membrane trafficking.
Figures

References
Electronic-Database Information
-
- 3D-PSSM server, http://www.sbg.bio.ic.ac.uk/servers/3dpssm/
-
- ClustalW, http://www.ebi.ac.uk/clustalw/
-
- Ensembl Genome database, http://www.ensembl.org/ (for mouse VAP [accession number ENSMUSP00000029026], rat VAP [accession number ENSRNOP00000007554], human VAPA [accession number ENSP00000217602], and human VAPB [accession number ENSP00000265619])
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for D. melanogaster DVAP-33A [accession number AY060395], S. cerevisiae SCS2 [accession number P40075], and A. californica VAP-33 [accession number Q16943])
-
- MapView at the National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/mapview/
References
-
- Chen Y-Z, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, Nicholson GA, Auer-Grumbach M, Wagner K, De Jonghe P, Griffin JW, Fischbeck KH, Timmerman V, Cornblath DR, Chance PF (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74:1128–1135 - PMC - PubMed
-
- Corcia P, Khoris J, Couratier P, Mayeux-Portas V, Bieth E, De Toffol B, Autret A, et al (2002) SMN1 gene study in three families in which ALS and spinal muscular atrophy coexist. Neurology 59:1464–1466 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
