

Long-term Safety and Efficacy of Avalglucosidase Alfa in Patients With Late-Onset Pompe Disease
- PMID: 35618441
- PMCID: PMC9421599
- DOI: 10.1212/WNL.0000000000200746
Long-term Safety and Efficacy of Avalglucosidase Alfa in Patients With Late-Onset Pompe Disease
Abstract
Background and objectives: Pompe disease is a rare, progressive neuromuscular disorder caused by deficiency of lysosomal acid α-glucosidase (GAA) and subsequent glycogen accumulation. Avalglucosidase alfa, a recombinant human GAA enzyme replacement therapy designed for increased cellular uptake and glycogen clearance, has been studied for long-term efficacy and safety in patients with late-onset Pompe disease (LOPD). Here, we report up to 6.5 years' experience with avalglucosidase alfa during the NEO1 and NEO-EXT studies.
Methods: NEO1 participants with LOPD, either treatment naive (Naive Group) or receiving alglucosidase alfa for ≥9 months (Switch Group), received avalglucosidase alfa (5, 10, or 20 mg/kg every other week [qow]) for 6 months before entering NEO-EXT and continued their NEO1 dose until all proceeded with 20 mg/kg qow. Safety and efficacy, a prespecified exploratory secondary outcome, were assessed; slopes of change for efficacy outcomes were calculated from a repeated mixed-measures model.
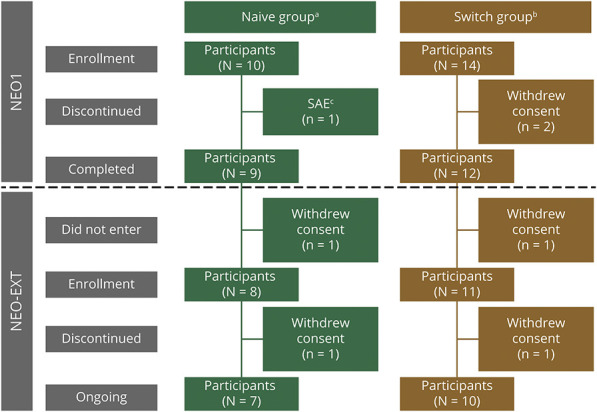
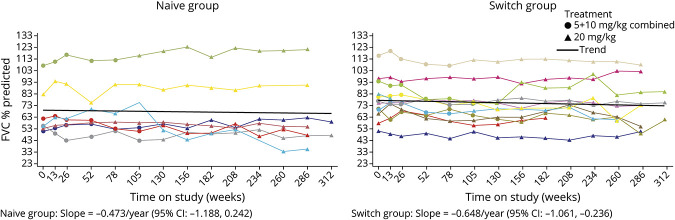
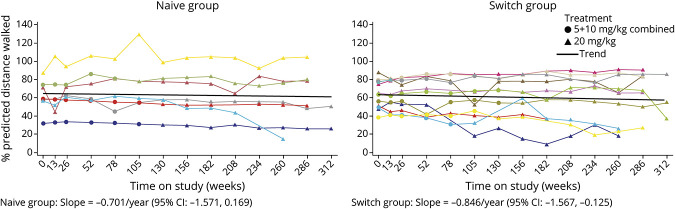
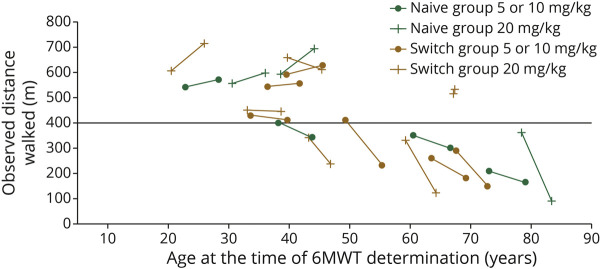
Results: Twenty-four participants enrolled in NEO1 (Naive Group, n = 10; Switch Group, n = 14); 21 completed and 19 entered NEO-EXT; in February 2020, 17 participants remained in NEO-EXT, with data up to 6.5 years. Avalglucosidase alfa was generally well tolerated during NEO-EXT, with a safety profile consistent with that in NEO1. No deaths or treatment-related life-threatening serious adverse events occurred. Eighteen participants developed antidrug antibodies without apparent effect on clinical outcomes. No participants who were tested developed immunoglobulin E antibodies. Upright forced vital capacity %predicted remained stable in most participants, with slope estimates (95% CIs) of -0.473 per year (-1.188 to 0.242) and -0.648 per year (-1.061 to -0.236) in the Naive and Switch Groups, respectively. Six-minute walk test (6MWT) %predicted was also stable for most participants, with slope estimates of -0.701 per year (-1.571 to 0.169) and -0.846 per year (-1.567 to -0.125) for the Naive and Switch Groups, respectively. Improvements in 6MWT distance were observed in most participants aged <45 years at NEO1 enrollment in both the Naive and Switch Groups.
Discussion: Avalglucosidase alfa was generally well tolerated for up to 6.5 years in adult participants with LOPD either naive to alglucosidase alfa or who had previously received alglucosidase alfa for ≥9 months.
Classification of evidence: This study provides Class IV evidence of long-term tolerability and sustained efficacy of avalglucosidase alfa in patients with LOPD after up to 6.5 years.
Trial registration information: NCT01898364 (NEO1 first posted: July 12, 2013; clinicaltrials.gov/ct2/show/NCT01898364); NCT02032524 (NEO-EXT first posted: January 10, 2014; clinicaltrials.gov/ct2/show/NCT02032524). First participant enrollment: NEO1-August 19, 2013; NEO-EXT-February 27, 2014.
Copyright © 2022 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
Similar articles
-
Enzyme replacement therapy for late-onset Pompe disease.Cochrane Database Syst Rev. 2023 Dec 12;12(12):CD012993. doi: 10.1002/14651858.CD012993.pub2. Cochrane Database Syst Rev. 2023. PMID: 38084761 Free PMC article.
-
The Mini-COMET Clinical Trial: Safety and Efficacy of Avalglucosidase Alfa after 97 Weeks of Treatment in Children with Infantile-Onset Pompe Disease Previously Treated with Alglucosidase Alfa.J Pediatr. 2025 Oct;285:114664. doi: 10.1016/j.jpeds.2025.114664. Epub 2025 May 29. J Pediatr. 2025. PMID: 40449831 Clinical Trial.
-
Efficacy and safety of avalglucosidase alfa in patients with late-onset Pompe disease after 145 weeks of treatment during the COMET trial.J Neurol. 2025 Aug 16;272(9):581. doi: 10.1007/s00415-025-13266-y. J Neurol. 2025. PMID: 40817977 Free PMC article. Clinical Trial.
-
An Indirect Treatment Comparison of Avalglucosidase Alfa versus Cipaglucosidase Alfa Plus Miglustat in Patients with Late-Onset Pompe Disease.Adv Ther. 2025 Sep 8. doi: 10.1007/s12325-025-03301-9. Online ahead of print. Adv Ther. 2025. PMID: 40920287
-
Enzyme replacement therapy for infantile-onset Pompe disease.Cochrane Database Syst Rev. 2017 Nov 20;11(11):CD011539. doi: 10.1002/14651858.CD011539.pub2. Cochrane Database Syst Rev. 2017. PMID: 29155436 Free PMC article.
Cited by
-
Efficacy of Switching Therapy From Alglucosidase Alfa to Avalglucosidase Alfa on Respiratory Function in Participants With Late-Onset Pompe Disease: A Post Hoc Analysis From the COMET Trial.JIMD Rep. 2025 Aug 12;66(5):e70033. doi: 10.1002/jmd2.70033. eCollection 2025 Sep. JIMD Rep. 2025. PMID: 40799512 Free PMC article.
-
Cardiovascular involvement in glycogen storage diseases.Nat Rev Cardiol. 2025 Jun 5. doi: 10.1038/s41569-025-01171-w. Online ahead of print. Nat Rev Cardiol. 2025. PMID: 40473899 Review.
-
Start, switch and stop (triple-S) criteria for enzyme replacement therapy of late-onset Pompe disease: European Pompe Consortium recommendation update 2024.Eur J Neurol. 2024 Sep;31(9):e16383. doi: 10.1111/ene.16383. Epub 2024 Jun 14. Eur J Neurol. 2024. PMID: 38873957 Free PMC article.
-
Enzyme replacement therapies in adults with Pompe disease: from trials to real-world data.Curr Opin Neurol. 2025 Oct 1;38(5):538-545. doi: 10.1097/WCO.0000000000001385. Epub 2025 Jun 5. Curr Opin Neurol. 2025. PMID: 40471681 Free PMC article. Review.
-
Efficacy of avalglucosidase alfa on forced vital capacity percent predicted in treatment-naïve patients with late-onset Pompe disease: A pooled analysis of clinical trials.Mol Genet Metab Rep. 2024 Jun 26;40:101109. doi: 10.1016/j.ymgmr.2024.101109. eCollection 2024 Sep. Mol Genet Metab Rep. 2024. PMID: 39035044 Free PMC article.
References
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous