

Genomic perspective on the bacillus causing paratyphoid B fever
- PMID: 39658567
- PMCID: PMC11632088
- DOI: 10.1038/s41467-024-54418-4
Genomic perspective on the bacillus causing paratyphoid B fever
Abstract
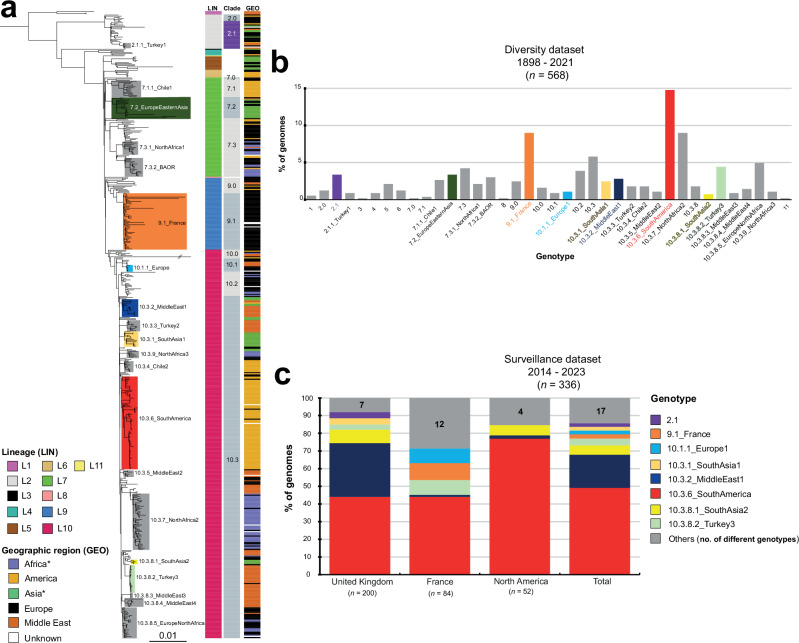
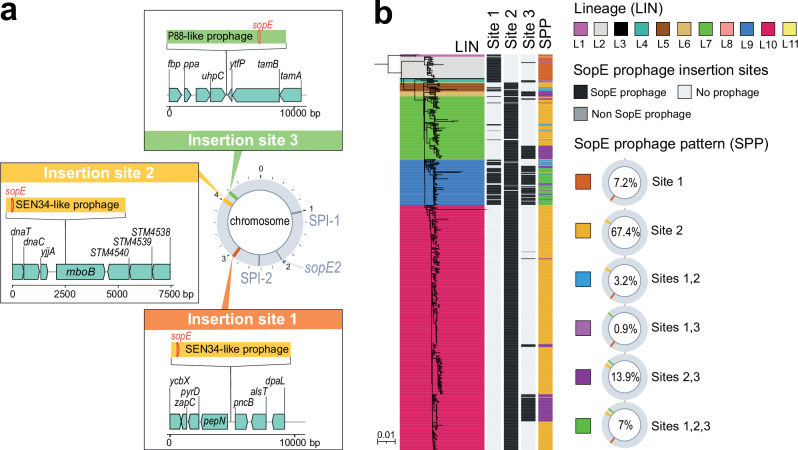
Paratyphoid B fever (PTB) is caused by an invasive lineage (phylogroup 1, PG1) of Salmonella enterica serotype Paratyphi B (SPB). However, little was known about the global population structure, geographic distribution, and evolution of this pathogen. Here, we report a whole-genome analysis of 568 historical and contemporary SPB PG1 isolates, obtained globally, between 1898 and 2021. We show that this pathogen existed in the 13th century, subsequently diversifying into 11 lineages and 38 genotypes with strong phylogeographic patterns. Following its discovery in 1896, it circulated across Europe until the 1970s, after which it was mostly reimported into Europe from South America, the Middle East, South Asia, and North Africa. Antimicrobial resistance recently emerged in various genotypes of SPB PG1, mostly through mutations of the quinolone-resistance-determining regions of gyrA and gyrB. This study provides an unprecedented insight into SPB PG1 and essential genomic tools for identifying and tracking this pathogen, thereby facilitating the global genomic surveillance of PTB.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
-
- Achard, C. & Bensaude, R. Infections paratyphoïdiques. Bull. Mem. Soc. Hop. Paris13, 820–833 (1896).
-
- Gwyn, N. B. On infection with a para-colon bacillus in a case with all the clinical features of typhoid fever. Bull. Johns. Hopkins Hosp.9, 54–56 (1898).
-
- Hirschfeld, L. A new germ of paratyphoid. Lancet193, 296–297 (1919).
-
- Pratt, J. H. On paratyphoid fever and its complications. Boston Med. Surg.148, 137–142 (1903).
-
- Proescher, F. & Roddy, J. A. Bacteriological studies on paratyphoid A and paratyphoid B. Arch. Intern. Med.3, 263–312 (1910).
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
