

Comparative genomic analysis of 60 Mycobacteriophage genomes: genome clustering, gene acquisition, and gene size
- PMID: 20064525
- PMCID: PMC2830324
- DOI: 10.1016/j.jmb.2010.01.011
Comparative genomic analysis of 60 Mycobacteriophage genomes: genome clustering, gene acquisition, and gene size
Abstract
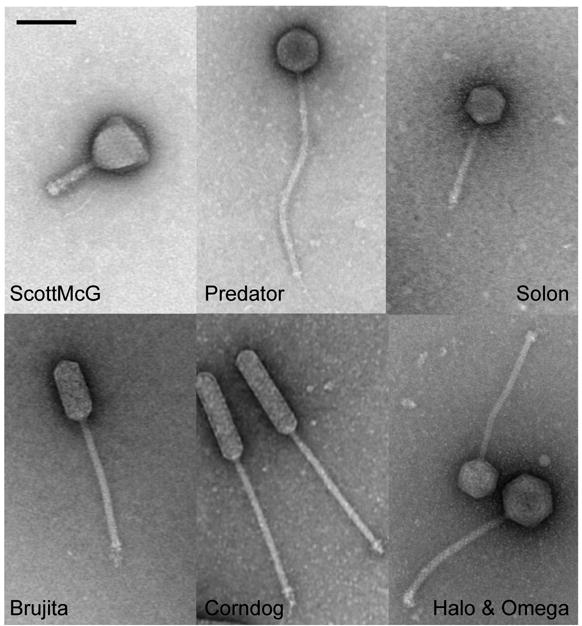
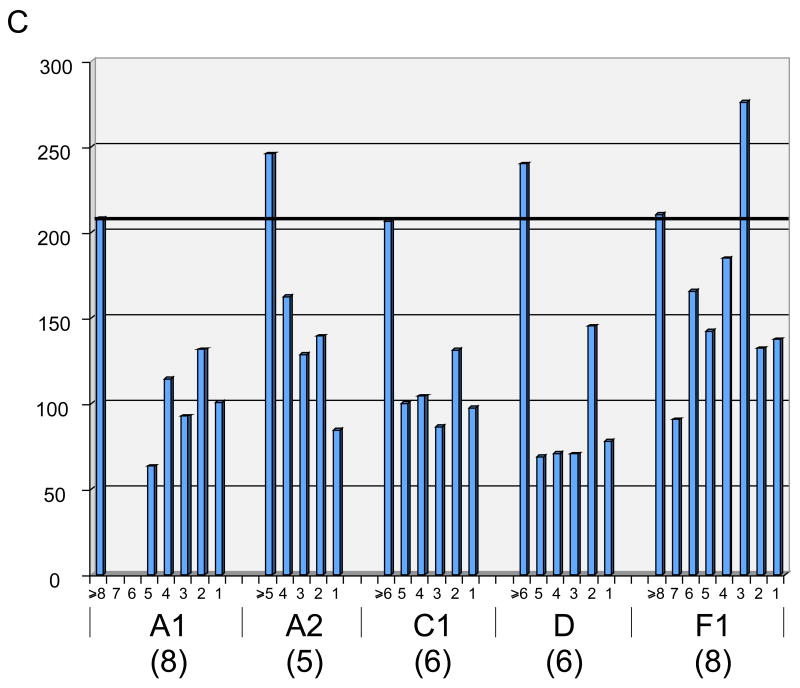
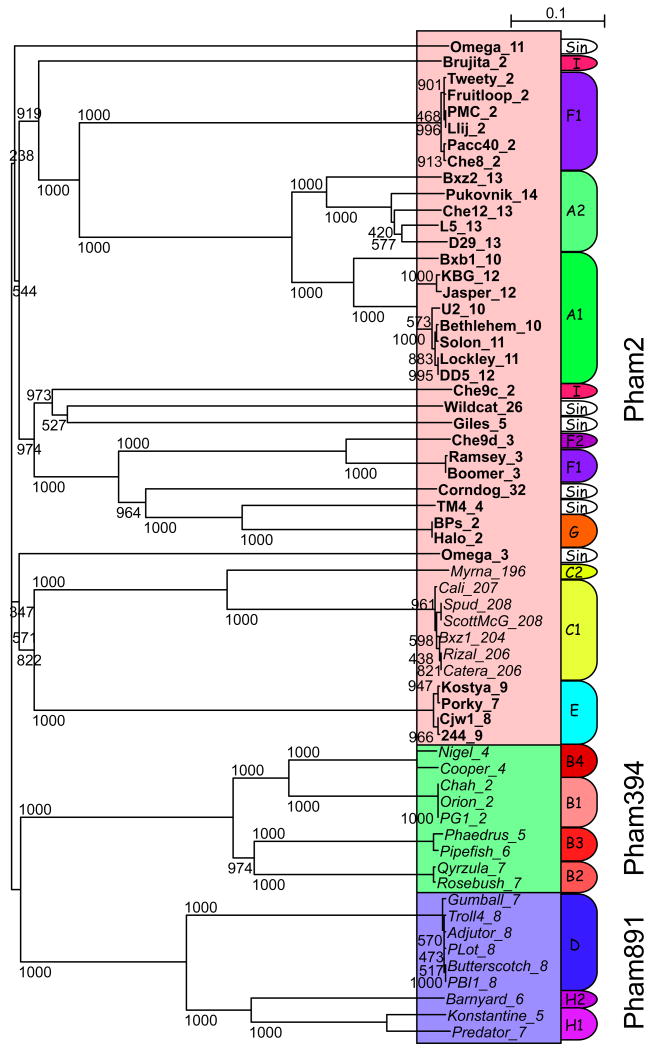
Mycobacteriophages are viruses that infect mycobacterial hosts. Expansion of a collection of sequenced phage genomes to a total of 60-all infecting a common bacterial host-provides further insight into their diversity and evolution. Of the 60 phage genomes, 55 can be grouped into nine clusters according to their nucleotide sequence similarities, 5 of which can be further divided into subclusters; 5 genomes do not cluster with other phages. The sequence diversity between genomes within a cluster varies greatly; for example, the 6 genomes in Cluster D share more than 97.5% average nucleotide similarity with one another. In contrast, similarity between the 2 genomes in Cluster I is barely detectable by diagonal plot analysis. In total, 6858 predicted open-reading frames have been grouped into 1523 phamilies (phams) of related sequences, 46% of which possess only a single member. Only 18.8% of the phams have sequence similarity to non-mycobacteriophage database entries, and fewer than 10% of all phams can be assigned functions based on database searching or synteny. Genome clustering facilitates the identification of genes that are in greatest genetic flux and are more likely to have been exchanged horizontally in relatively recent evolutionary time. Although mycobacteriophage genes exhibit a smaller average size than genes of their host (205 residues compared with 315), phage genes in higher flux average only 100 amino acids, suggesting that the primary units of genetic exchange correspond to single protein domains.
Copyright (c) 2010 Elsevier Ltd. All rights reserved.
Figures













References
-
- Hendrix RW. Bacteriophages: evolution of the majority. Theor Popul Biol. 2002;61:471–80. - PubMed
-
- Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR, Hendrix RW, Hatfull GF. Origins of highly mosaic mycobacteriophage genomes. Cell. 2003;113:171–82. - PubMed
-
- Brussow H, Hendrix RW. Phage genomics: small is beautiful. Cell. 2002;108:13–6. - PubMed
-
- Hendrix RW. Bacteriophage genomics. Curr Opin Microbiol. 2003;6:506–11. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
