

Adult genomic medicine: lessons from a multisite study of 2700 patients
- PMID: 41024252
- PMCID: PMC12477811
- DOI: 10.1186/s13073-025-01529-2
Adult genomic medicine: lessons from a multisite study of 2700 patients
Abstract
Background: Clinical exome and genome sequencing has transformed the diagnostic workup of patients with genetic disorders. The extensive body of evidence supporting the application of this clinical genomics approach in pediatric patients stands in stark contrast to the relative paucity of evidence for its use in the adult population. Here, we describe the largest cohort to date of adult patients who underwent clinical exome and genome sequencing for suspected genetic diagnoses.
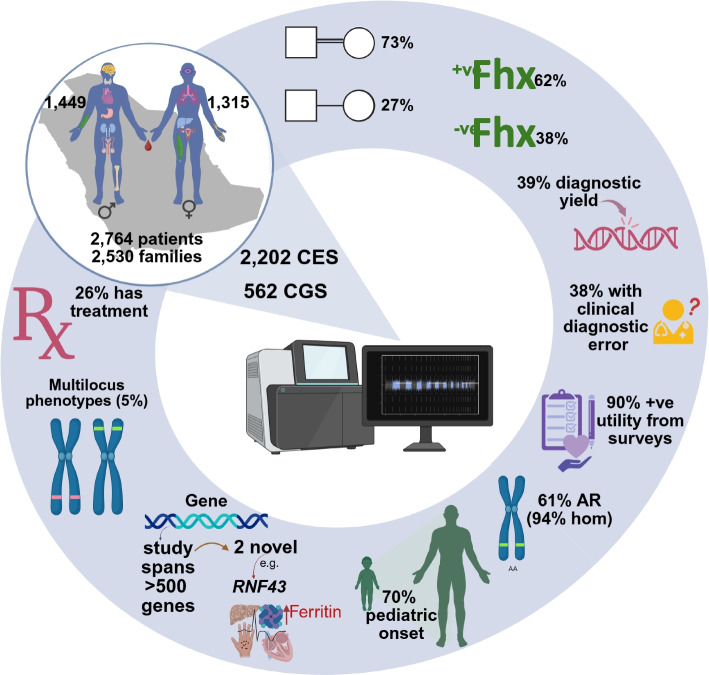
Methods: A total of 2763 adult patients (2529 families) from all regions of Saudi Arabia are included in this cohort (2202 exomes, and 561 genomes).
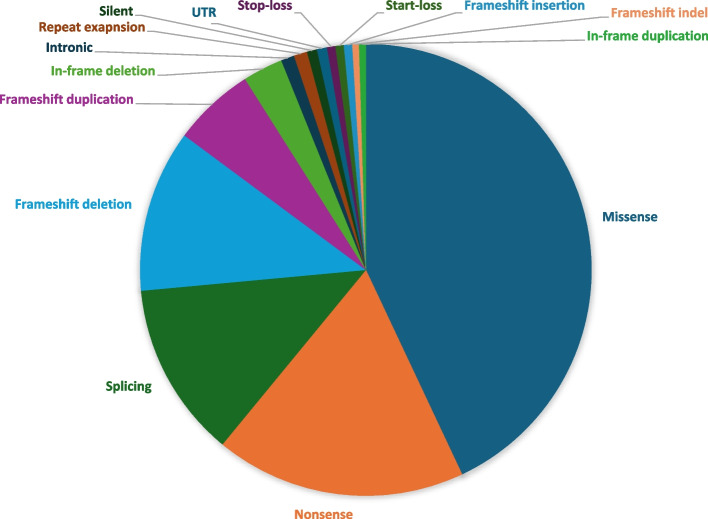
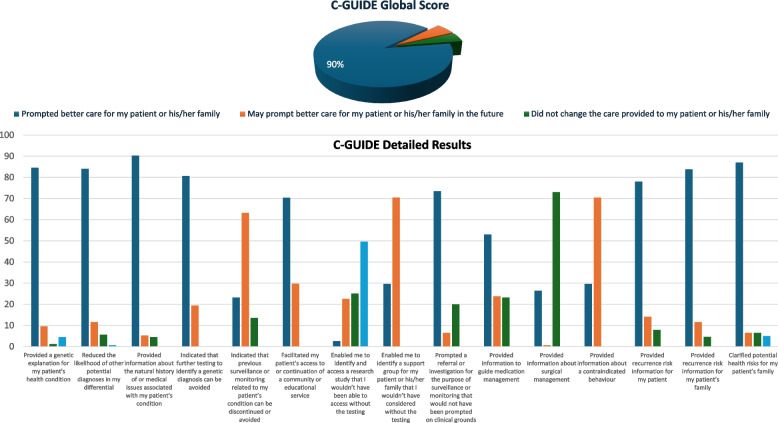
Results: The diagnostic rate is 38.9% spanning 535 Mendelian genes and revealing clinical diagnostic errors in 38% of patients with positive reports. Structured feedback using C-GUIDE demonstrates clinical utility in 90% of positive cases. Consistent with the highly consanguineous nature of the local population, the majority (61%) of diagnosed phenotypes are recessive (94.6% homozygous) and founder variants account for 85% (414/487) of these variants. The same population characteristic has also led to the encounter of extremely rare, even novel recessive disorders including a highly penetrant novel RNF43-related hemochromatosis, NFXL1-related syndrome of hyperlaxity, short stature, and kidney disease, as well as autosomal recessive forms of typically dominant disorders. Multilocus phenotypes are observed in 5% of cases although only 26.7% of these are caused by two recessive variants. That 70% of molecular diagnoses encountered in our cohort are typically described in pediatric patients allowed us to observe highly unusual clinical presentations in the adult population. This delayed diagnosis also represents a missed opportunity for effective treatment in many instances and we note the availability of treatment for 26% of diagnosed conditions. Of particular interest are patients with monogenic disorders that could be overlooked as common multifactorial adult diseases (e.g., diabetes, dyslipidemia, stroke, chronic kidney disease, and dementia). Finally, we note the opportunities of deploying adult clinical genomics in an underrepresented population where 45.5% (373/819) of encountered variants are completely absent in gnomAD.
Conclusions: Our results illustrate numerous benefits of a clinical genomics approach in adult medicine and argue for a broader implementation than currently practiced.
Keywords: Clinical utility; Exome; Founder variants; Mendelian phenocopies; Novel allelic disorders; Novel disease genes; Rare diseases; Reflex genome; Underrepresented population.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study adheres to the principles of the Declaration of Helsinki (1996) and was approved by the IRB with a waiver of consent by the Ethics Committee at the Office of Research Affairs, King Faisal Specialist Hospital, and Research Center (KFSHRC RAC# 2230016) for the use of aggregated data. Ethical approval was also granted by Ministry of National Guard Health Affairs (MNG-HA) Institutional Review Board (KAIMRC RC19/120/R). Consent for publication: Written informed consent was obtained from participants and/or their legal guardians for the publication of possible identifiable clinical details and identifiable facial images, in accordance with IRB-approved protocols (KFSHRC RAC# 2080006 and KAIMRC RC19/120/R). Competing interests: KB, HH, BA, AA, MA, WB and FSA are paid employees and TB is a paid consultant of Lifera Omics.
Figures


References
-
- Shickh S, Mighton C, Uleryk E, Pechlivanoglou P, Bombard Y. The clinical utility of exome and genome sequencing across clinical indications: a systematic review. Hum Genet. 2021;140(10):1403–16. - PubMed
-
- Guo F, Liu R, Pan Y, Collins C, Bean L, Ma Z, et al. Evidence from 2100 index cases supports genome sequencing as a first-tier genetic test. Genet Med. 2024;26(1):100995. - PubMed
-
- Chung CCY, Hue SPY, Ng NYT, Doong PHL, Hong Kong Genome Project, Chu ATW, et al. Meta-analysis of the diagnostic and clinical utility of exome and genome sequencing in pediatric and adult patients with rare diseases across diverse populations. Genet Med Off J Am Coll Med Genet. 2023;25(9):100896. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
