

Whole genome sequencing reveals novel IGHMBP2 variant leading to unique cryptic splice-site and Charcot-Marie-Tooth phenotype with early onset symptoms
- PMID: 31020813
- PMCID: PMC6565564
- DOI: 10.1002/mgg3.676
Whole genome sequencing reveals novel IGHMBP2 variant leading to unique cryptic splice-site and Charcot-Marie-Tooth phenotype with early onset symptoms
Abstract
Background: Rare variants (RV) in immunoglobulin mu-binding protein 2 (IGHMBP2) [OMIM 600502] can cause an autosomal recessive type of Charcot-Marie-Tooth (CMT) disease [OMIM 616155], an inherited peripheral neuropathy. Over 40 different genes are associated with CMT, with different possible inheritance patterns.
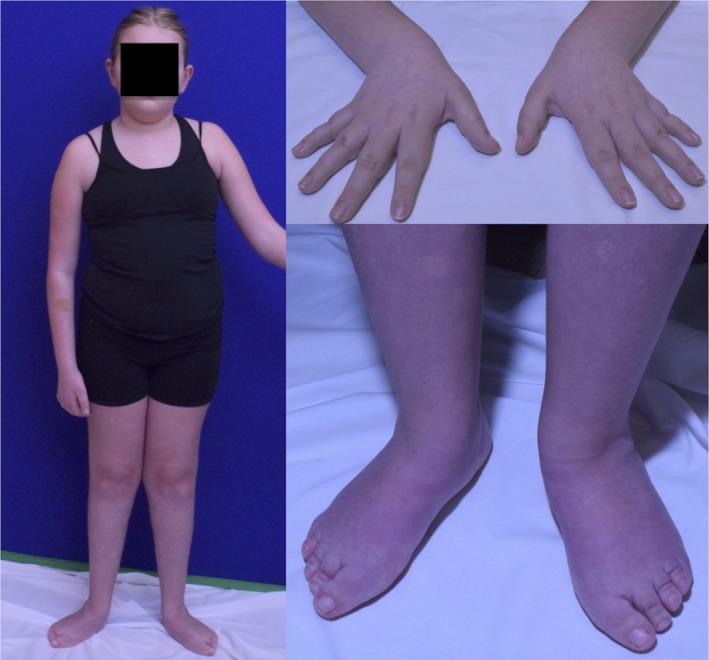
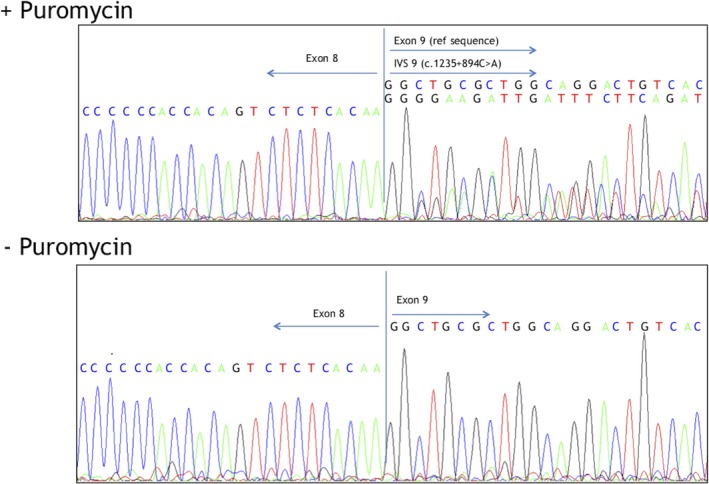
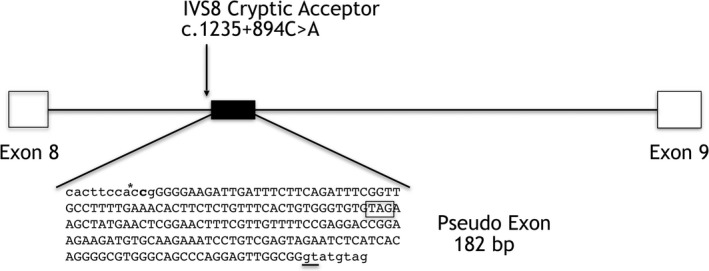
Methods and results: An 11-year-old female with motor delays was found to have distal atrophy, weakness, and areflexia without bulbar or sensory findings. Her clinical evaluation was unrevealing. Whole exome sequencing (WES) revealed a maternally inherited IGHMBP2 RV (c.1730T>C) predicted to be pathogenic, but no variant on the other allele was identified. Deletion and duplication analysis was negative. She was referred to the Undiagnosed Disease Network (UDN) for further evaluation. Whole genome sequencing (WGS) confirmed the previously identified IGHMBP2 RV and identified a paternally inherited non-coding IGHMBP2 RV. This was predicted to activate a cryptic splice site perturbing IGHMBP2 splicing. Reverse transcriptase polymerase chain reaction (RT-PCR) analysis was consistent with activation of the cryptic splice site. The abnormal transcript was shown to undergo nonsense-mediated decay (NMD), resulting in halpoinsufficiency.
Conclusion: This case demonstrates the deficiencies of WES and traditional molecular analyses and highlights the advantages of utilization of WGS and functional studies.
Keywords: IGHMBP2; Charcot-Marie-Tooth; Undiagnosed Disease Network; intron; splicing; whole exome sequencing.
© 2019 The Authors. Molecular Genetics & Genomic Medicine published by Wiley Periodicals, Inc.
Conflict of interest statement
The authors have no disclosures or conflicts of interest.
Figures
References
-
- Bird, T. D . (1993). Charcot‐Marie‐Tooth hereditary neuropathy overview. GeneReviews, 1–25. https://doi.org/nbk1358 [bookaccession] - PubMed
-
- Carss, K. J. , Arno, G. , Erwood, M. , Stephens, J. , Sanchis‐Juan, A. , Hull, S. , … Raymond, F. L. (2017). Comprehensive rare variant analysis via whole‐genome sequencing to determine the molecular pathology of inherited retinal disease. American Journal of Human Genetics, 100(1), 75–90. 10.1016/j.ajhg.2016.12.003 - DOI - PMC - PubMed
-
- Carter, M. S. , Doskow, J. , Morris, P. , Li, S. , Nhim, R. P. , Sandstedt, S. , & Wilkinson, M. F. (1995). A regulatory mechanism that detects premature nonsense codons in T‐cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. Journal of Biological Chemistry, 270(48), 28995–29003. 10.1074/jbc.270.48.28995 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
