

Best practices for differential accessibility analysis in single-cell epigenomics
- PMID: 39394227
- PMCID: PMC11470024
- DOI: 10.1038/s41467-024-53089-5
Best practices for differential accessibility analysis in single-cell epigenomics
Abstract
Differential accessibility (DA) analysis of single-cell epigenomics data enables the discovery of regulatory programs that establish cell type identity and steer responses to physiological and pathophysiological perturbations. While many statistical methods to identify DA regions have been developed, the principles that determine the performance of these methods remain unclear. As a result, there is no consensus on the most appropriate statistical methods for DA analysis of single-cell epigenomics data. Here, we present a systematic evaluation of statistical methods that have been applied to identify DA regions in single-cell ATAC-seq (scATAC-seq) data. We leverage a compendium of scATAC-seq experiments with matching bulk ATAC-seq or scRNA-seq in order to assess the accuracy, bias, robustness, and scalability of each statistical method. The structure of our experiments also provides the opportunity to define best practices for the analysis of scATAC-seq data beyond DA itself. We leverage this understanding to develop an R package implementing these best practices.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
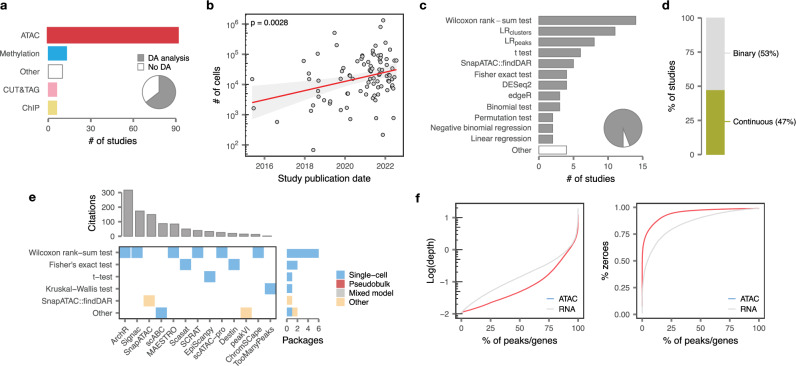


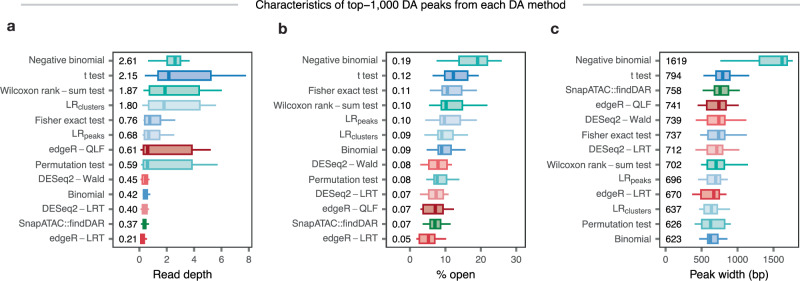

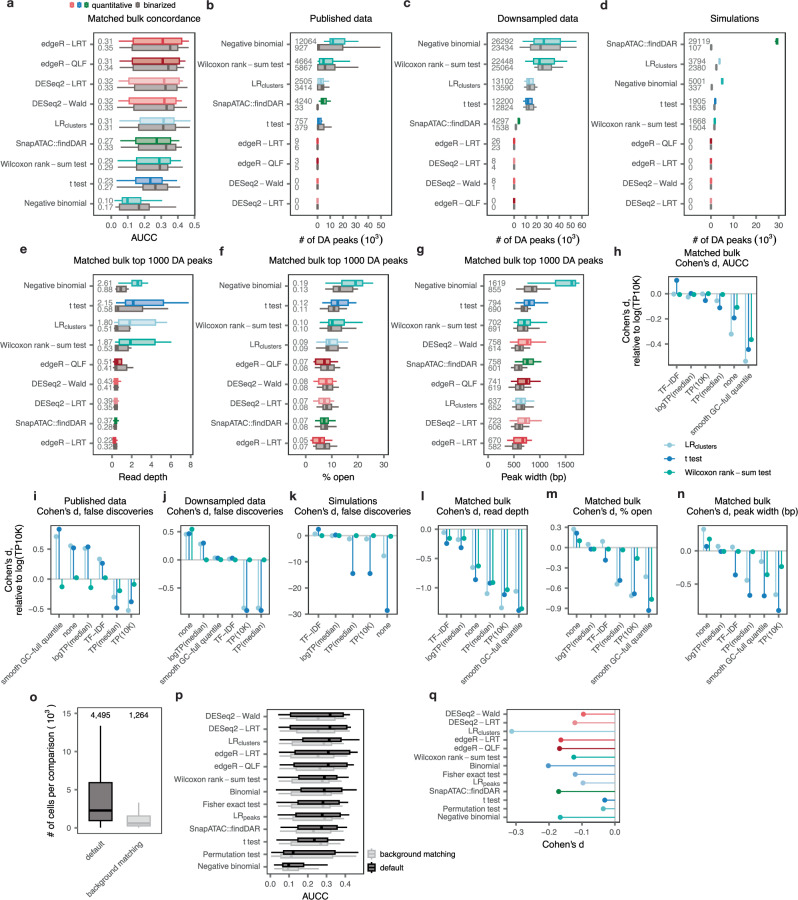

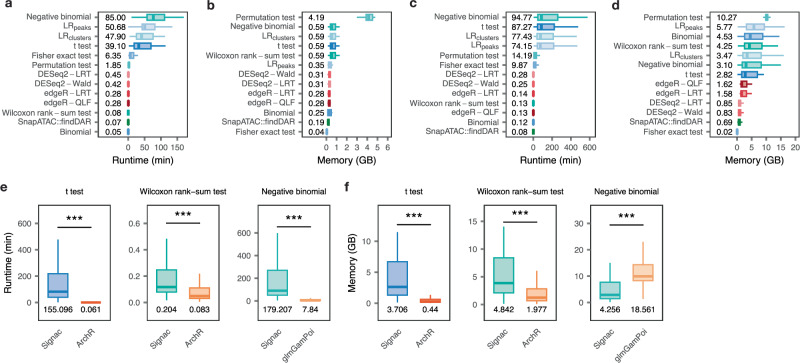

References
-
- Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet.20, 207–220 (2019). - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Research Materials
