

Canonical androgen response element motifs are tumor suppressive regulatory elements in the prostate
- PMID: 39672812
- PMCID: PMC11645413
- DOI: 10.1038/s41467-024-53734-z
Canonical androgen response element motifs are tumor suppressive regulatory elements in the prostate
Abstract
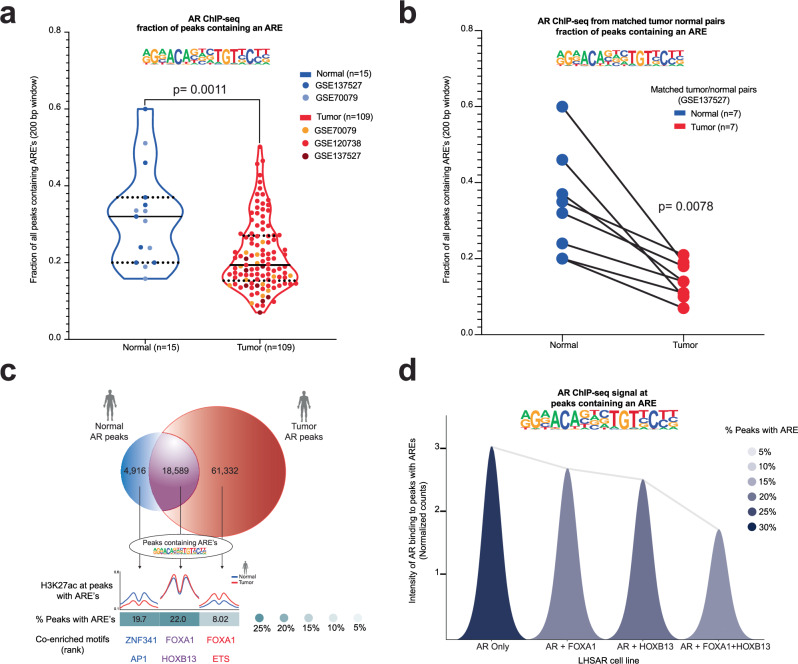
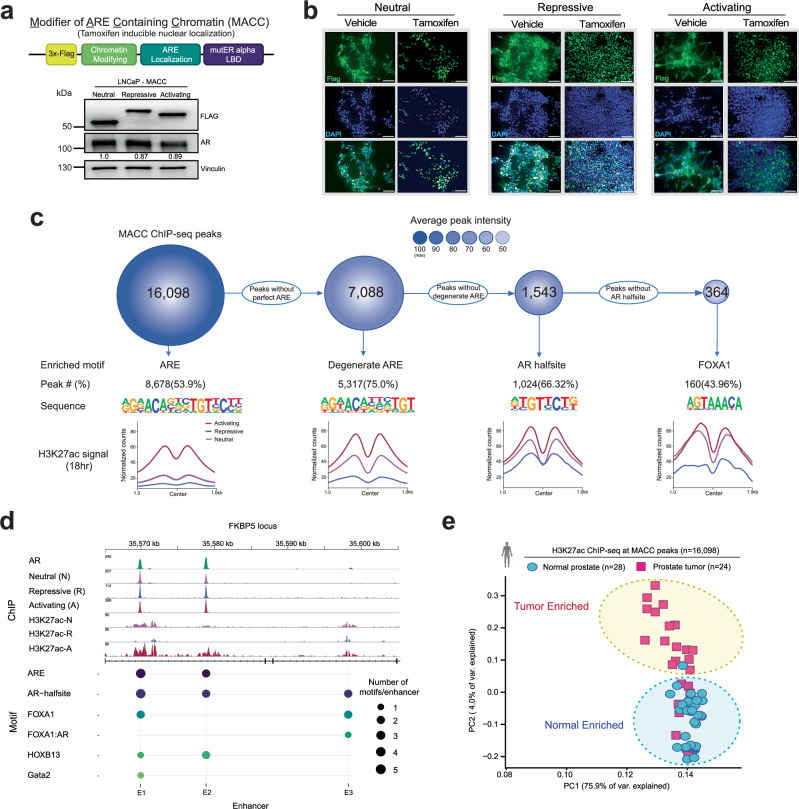
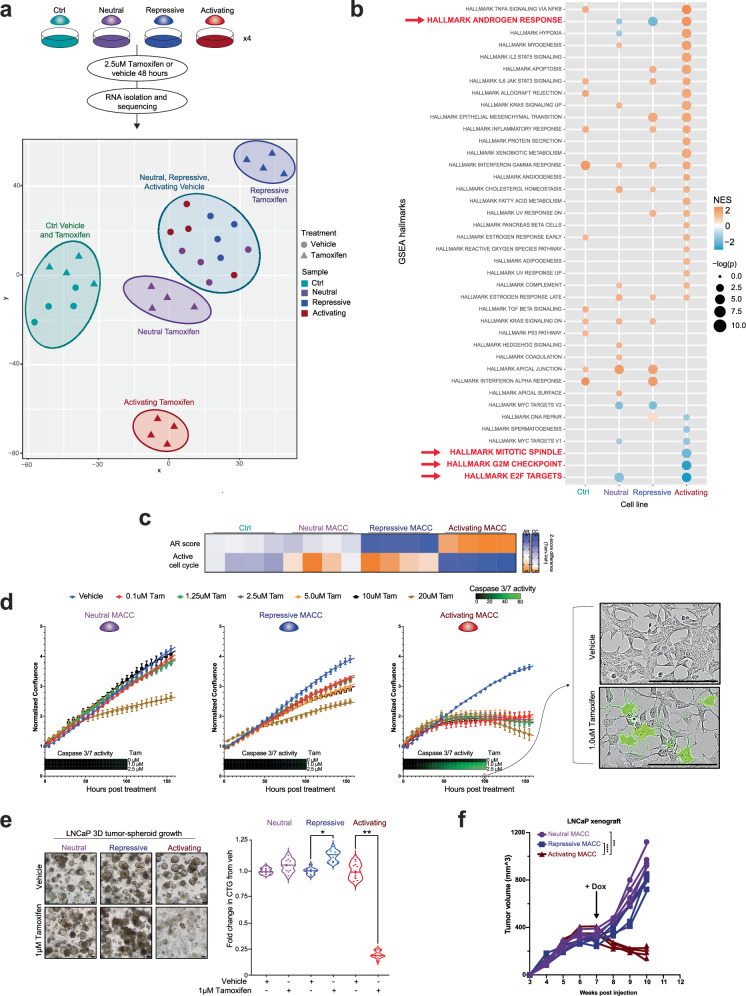
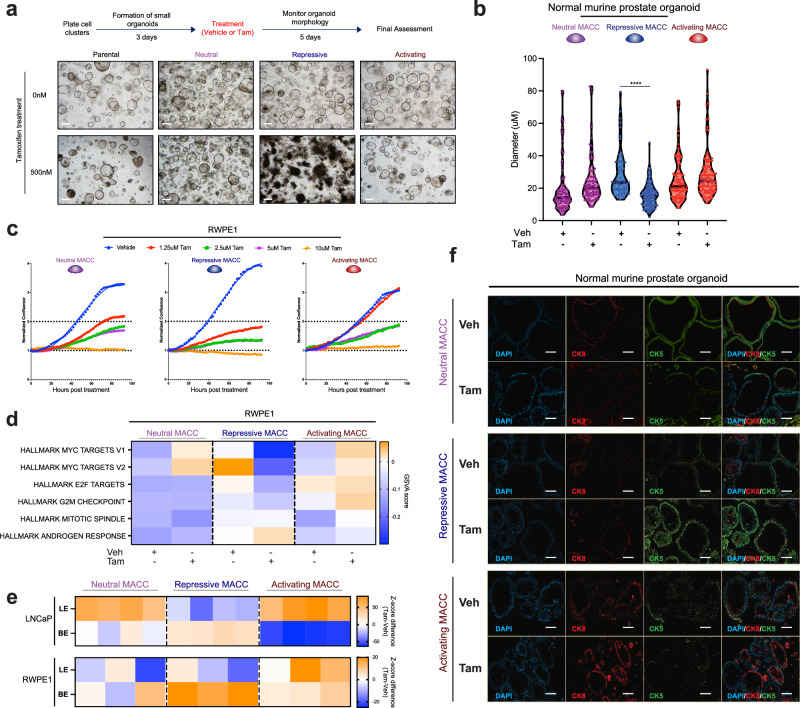
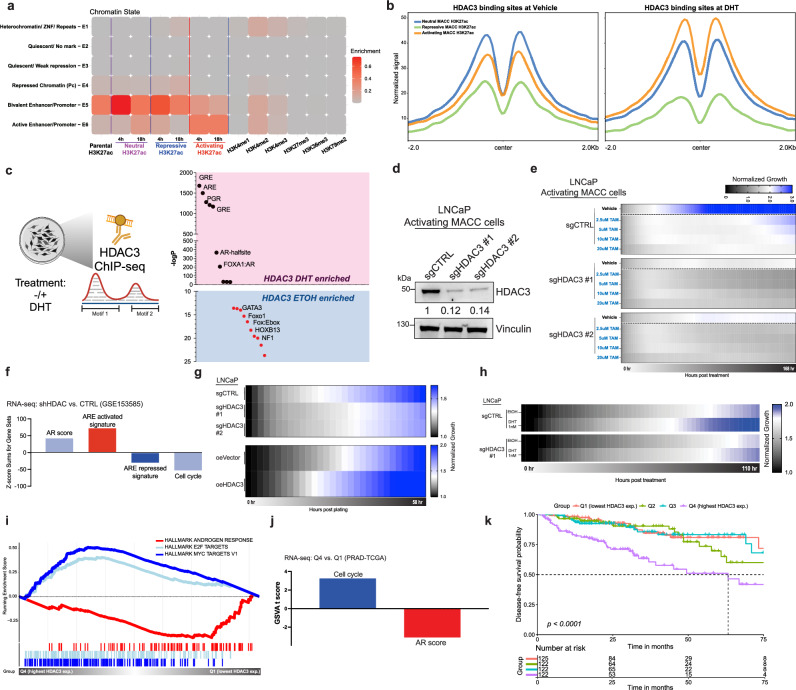
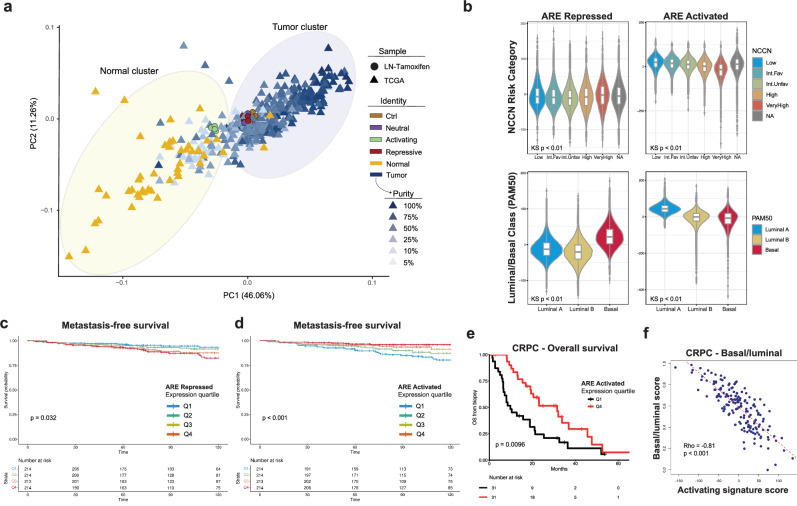
The androgen receptor (AR) is central in prostate tissue identity and differentiation, and controls normal growth-suppressive, prostate-specific gene expression. It also drives prostate tumorigenesis when hijacked for oncogenic transcription. The execution of growth-suppressive AR transcriptional programs in prostate cancer (PCa) and the potential for reactivation remain unclear. Here, we use a genome-wide approach to modulate canonical androgen response element (ARE) motifs-the classic DNA binding elements for AR-to delineate distinct AR transcriptional programs. We find that activating these AREs promotes differentiation and growth-suppressive transcription, potentially leading to AR+ PCa cell death, while ARE repression is tolerated by PCa cells but deleterious to normal prostate cells. Gene signatures driven by ARE activity correlate with improved prognosis and luminal phenotypes in PCa patients. Canonical AREs maintain a normal, lineage-specific transcriptional program that can be reengaged in PCa cells, offering therapeutic potential and clinical relevance.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: M.S. reports grants from the Swedish Research Council, the Swedish Society of Medicine, and the Prostate Cancer Foundation during the conduct of the study. A.H., Y.L., and E.D. are employees of Veracyte, Inc. M.A.A. and D.L. are currently employees of Loxo Oncology. C.E.B. is a co-inventor on a patent issued to Weill Medical College of Cornell University on SPOP mutations in prostate cancer. F.Y.F. reports fees from Janssen Oncology, Bayer, PFS Genomics, Myovant Sciences, Roivant Sciences, Astellas Pharma, Foundation Medicine, Varian, Bristol Myers Squibb (BMS), Exact Sciences, BlueStar Genomics, Novartis, and Tempus; other support from Serimmune and Artera outside the submitted work. The authors declare no other potential competing interests.
Figures
Update of
-
Canonical AREs are tumor suppressive regulatory elements in the prostate.bioRxiv [Preprint]. 2024 Feb 27:2024.02.23.581466. doi: 10.1101/2024.02.23.581466. bioRxiv. 2024. Update in: Nat Commun. 2024 Dec 13;15(1):10675. doi: 10.1038/s41467-024-53734-z. PMID: 38464162 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
