

Allele-specific DNA methylation in mouse strains is mainly determined by cis-acting sequences
- PMID: 19687144
- PMCID: PMC2775599
- DOI: 10.1101/gr.095562.109
Allele-specific DNA methylation in mouse strains is mainly determined by cis-acting sequences
Abstract
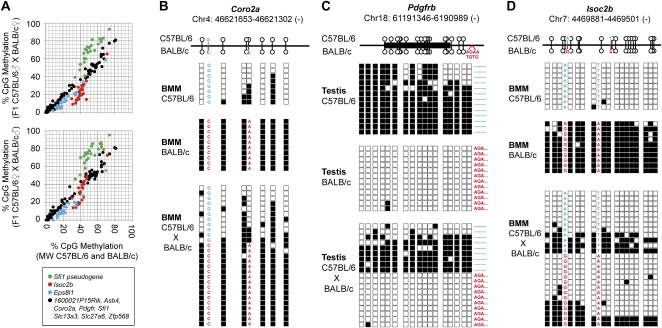
DNA methylation is a vital epigenetic mark that participates in establishing and maintaining chromatin structures and regulates gene transcription during mammalian development and cellular differentiation. Differences in epigenetic patterns between individuals may contribute to phenotypic variation and disease susceptibility; however, little is known about the extent of such variation or how different epigenetic patterns are established. Here we have compared DNA methylation profiles of macrophages from two inbred mouse strains (C57BL/6 and BALB/c) at 181 large genomic intervals that were selected based on differential gene expression patterns. Using a DNA methylation-dependent fractionation approach based on a combination of methyl-CpG immunoprecipitation and locus-wide tiling arrays, we identified several hundred differentially methylated regions, and simultaneously uncovered previously unrecognized genetic variability between both mouse strains at the studied loci. DNA sequence and methylation differences were validated by DNA sequencing and mass spectrometry analysis of bisulfite-treated DNA for a subset of regions. Importantly, we show that in F1 hybrids, the majority of strain-specific methylation patterns in somatic cells were maintained on the parental allele, regardless of their status in the male germ line. The common association of differentially methylated regions with sequence polymorphisms suggests that the genomic context determines the developmentally regulated epigenetic status at most nonimprinted regions of mammalian genomes.
Figures
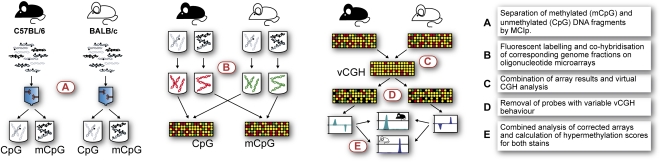
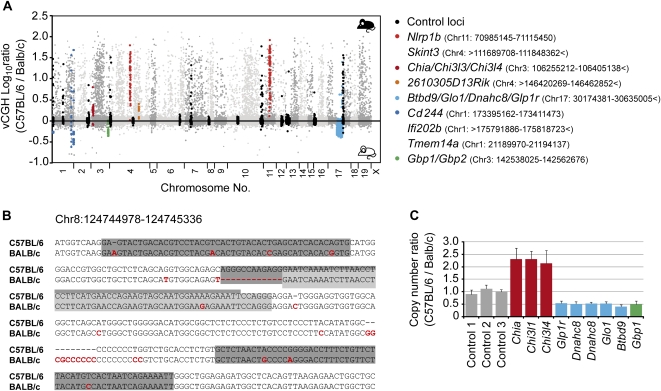
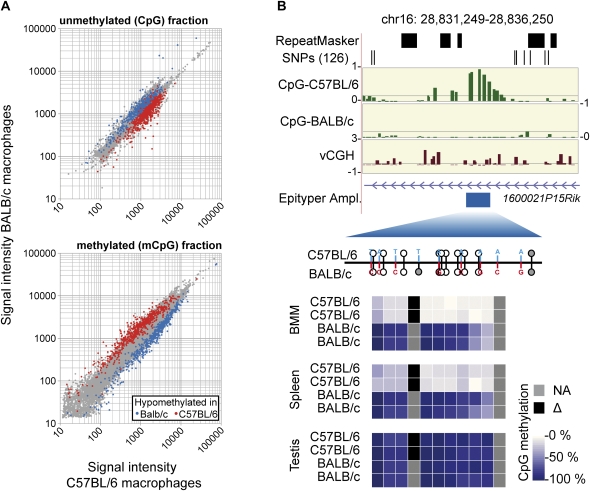
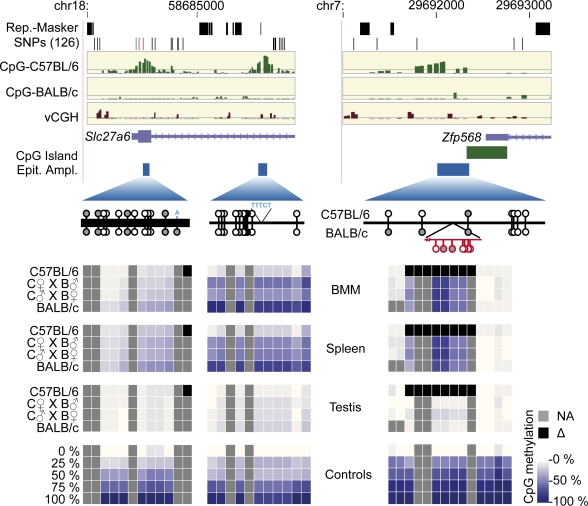
References
-
- Ashe A, Morgan DK, Whitelaw NC, Bruxner TJ, Vickaryous NK, Cox LL, Butterfield NC, Wicking C, Blewitt ME, Wilkins SJ, et al. A genome-wide screen for modifiers of transgene variegation identifies genes with critical roles in development. Genome Biol. 2008;9:R182. doi: 10.1186/gb-2008-9-12-r182. - DOI - PMC - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes & Dev. 2002;16:6–21. - PubMed
-
- Blewitt ME, Gendrel AV, Pang Z, Sparrow DB, Whitelaw N, Craig JM, Apedaile A, Hilton DJ, Dunwoodie SL, Brockdorff N, et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet. 2008;40:663–669. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases