

scConfluence: single-cell diagonal integration with regularized Inverse Optimal Transport on weakly connected features
- PMID: 39237488
- PMCID: PMC11377776
- DOI: 10.1038/s41467-024-51382-x
scConfluence: single-cell diagonal integration with regularized Inverse Optimal Transport on weakly connected features
Abstract
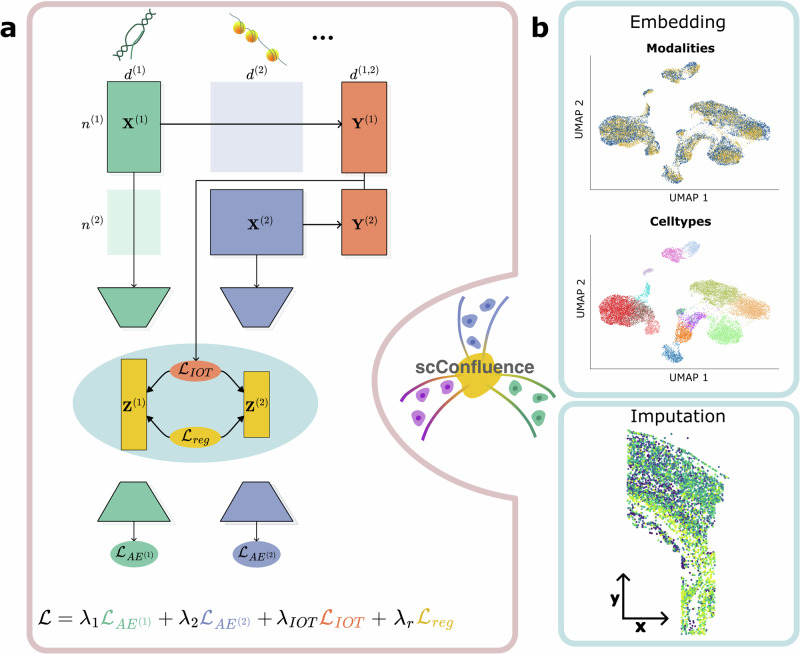
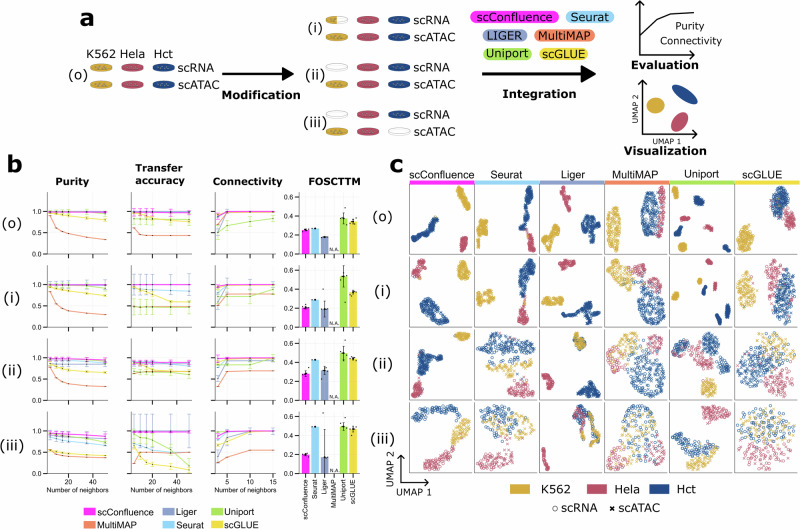
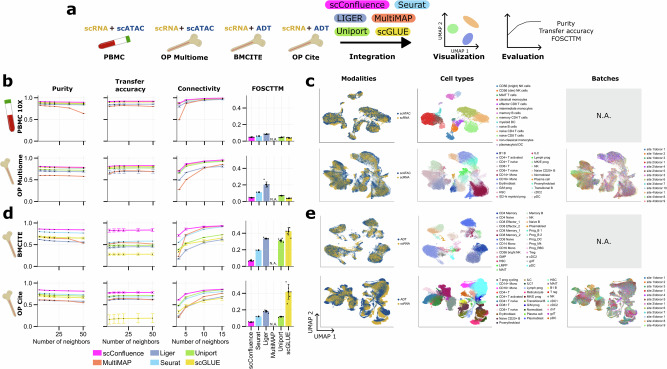
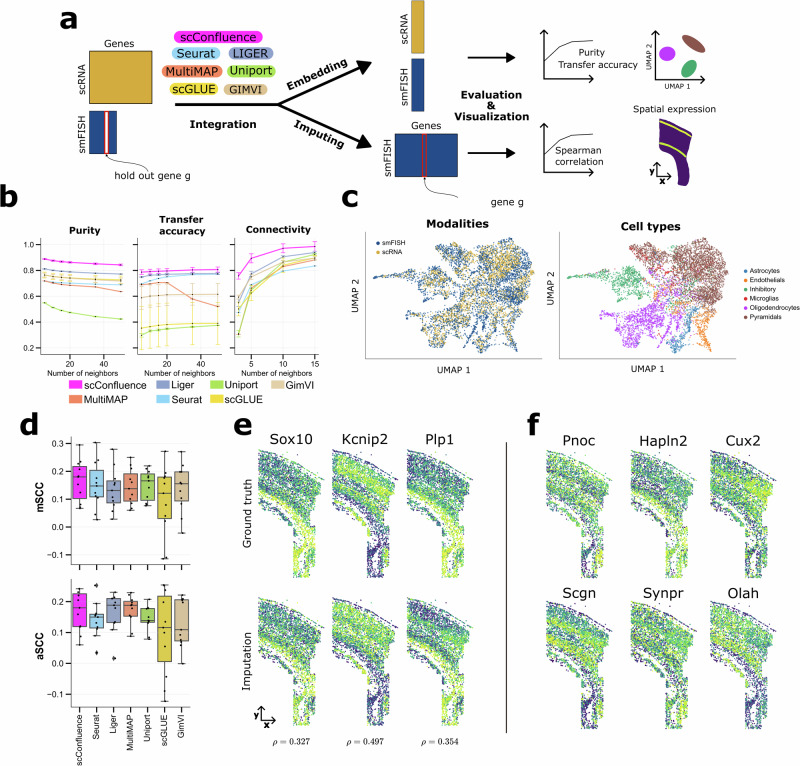
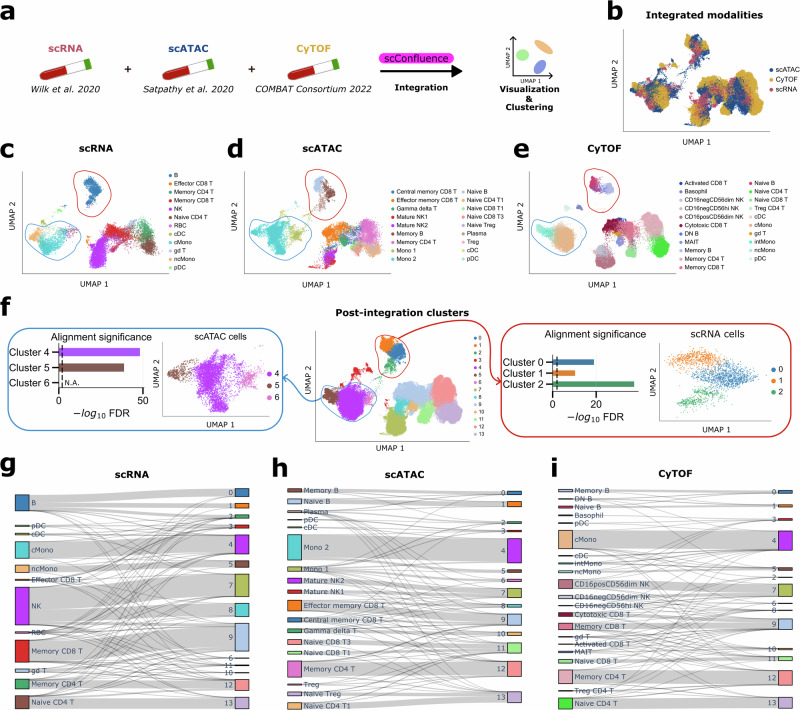
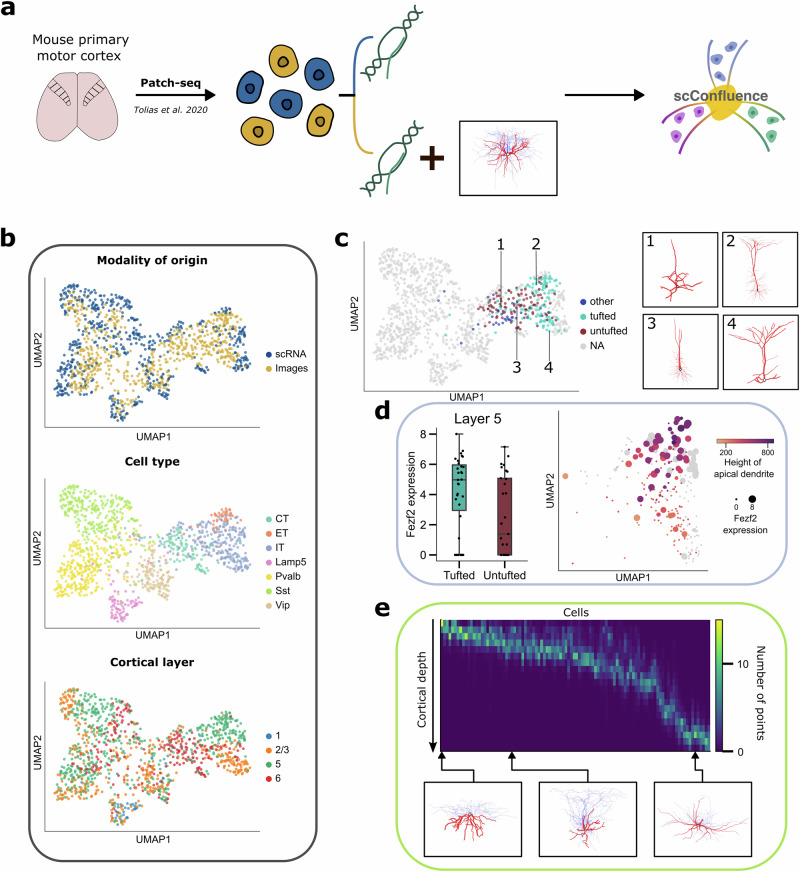
The abundance of unpaired multimodal single-cell data has motivated a growing body of research into the development of diagonal integration methods. However, the state-of-the-art suffers from the loss of biological information due to feature conversion and struggles with modality-specific populations. To overcome these crucial limitations, we here introduce scConfluence, a method for single-cell diagonal integration. scConfluence combines uncoupled autoencoders on the complete set of features with regularized Inverse Optimal Transport on weakly connected features. We extensively benchmark scConfluence in several single-cell integration scenarios proving that it outperforms the state-of-the-art. We then demonstrate the biological relevance of scConfluence in three applications. We predict spatial patterns for Scgn, Synpr and Olah in scRNA-smFISH integration. We improve the classification of B cells and Monocytes in highly heterogeneous scRNA-scATAC-CyTOF integration. Finally, we reveal the joint contribution of Fezf2 and apical dendrite morphology in Intra Telencephalic neurons, based on morphological images and scRNA.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Method of the Year 2013. Nat. Methods11, 1–1 (2014). - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
