

The Drosophila melanogaster transcriptome by paired-end RNA sequencing
- PMID: 21177959
- PMCID: PMC3032934
- DOI: 10.1101/gr.107854.110
The Drosophila melanogaster transcriptome by paired-end RNA sequencing
Abstract
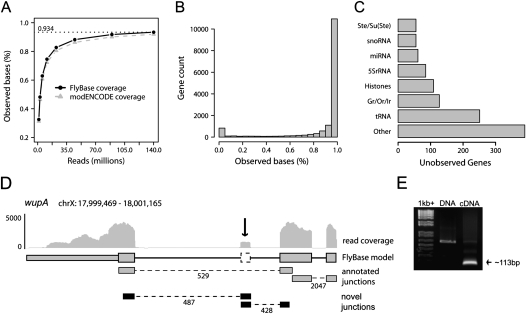
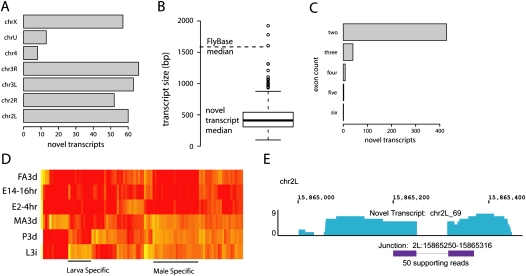
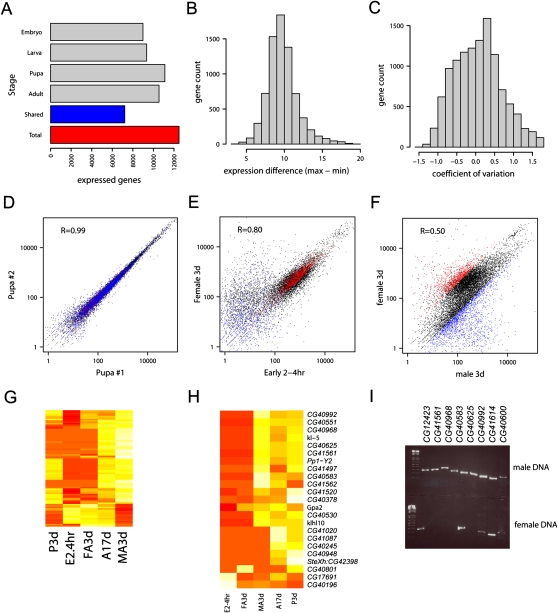
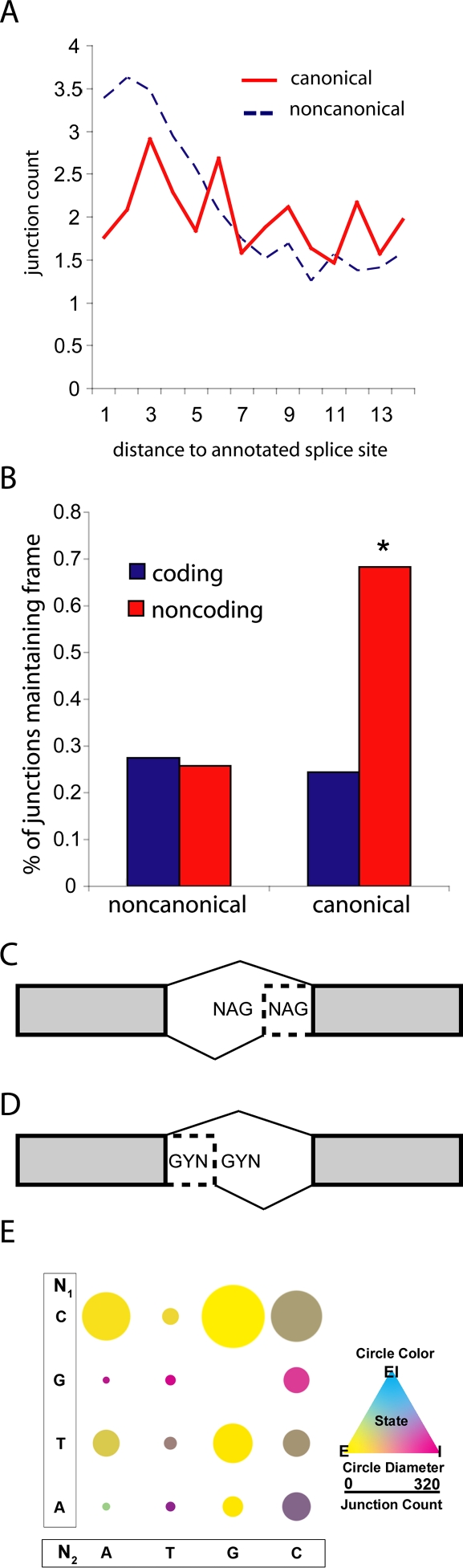
RNA-seq was used to generate an extensive map of the Drosophila melanogaster transcriptome by broad sampling of 10 developmental stages. In total, 142.2 million uniquely mapped 64-100-bp paired-end reads were generated on the Illumina GA II yielding 356× sequencing coverage. More than 95% of FlyBase genes and 90% of splicing junctions were observed. Modifications to 30% of FlyBase gene models were made by extension of untranslated regions, inclusion of novel exons, and identification of novel splicing events. A total of 319 novel transcripts were identified, representing a 2% increase over the current annotation. Alternate splicing was observed in 31% of D. melanogaster genes, a 38% increase over previous estimations, but significantly less than that observed in higher organisms. Much of this splicing is subtle such as tandem alternate splice sites.
Figures
References
-
- Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP 2002. Gene expression during the life cycle of Drosophila melanogaster. Science 297: 2270–2275 - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases