

Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes
- PMID: 22056041
- PMCID: PMC3225288
- DOI: 10.1016/j.cell.2011.10.002
Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes
Abstract
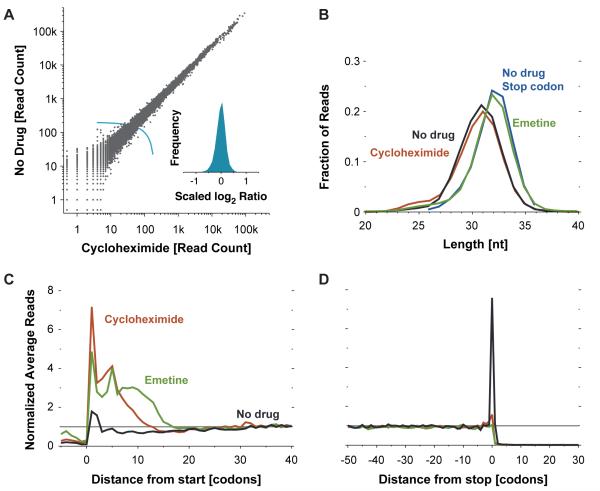
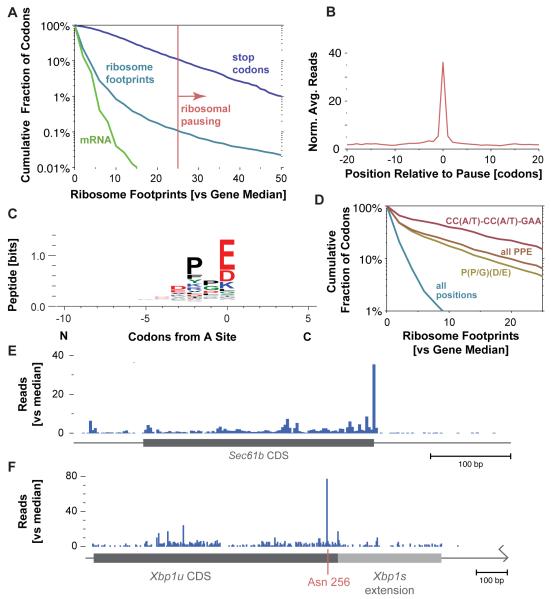
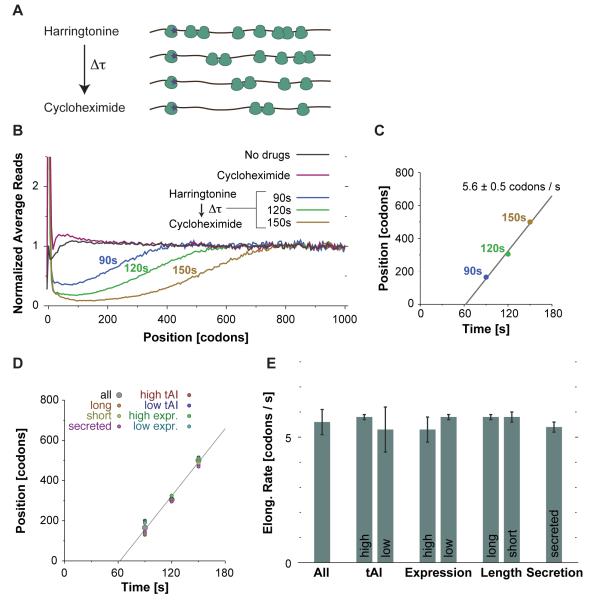
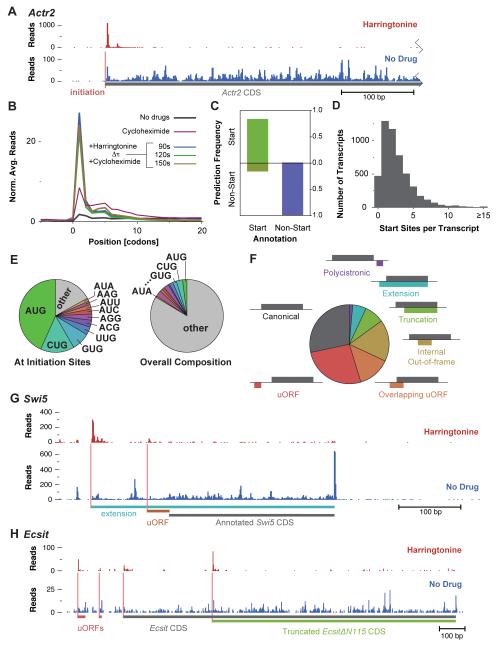
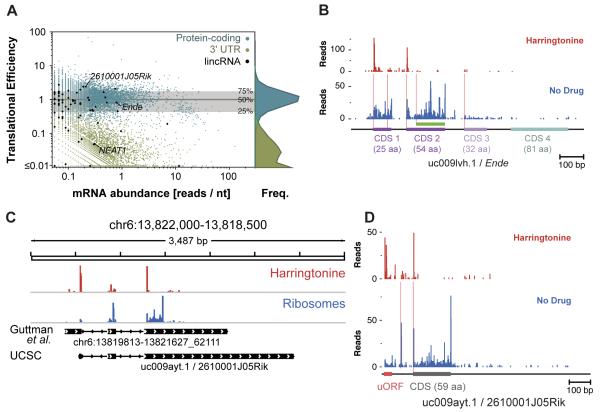
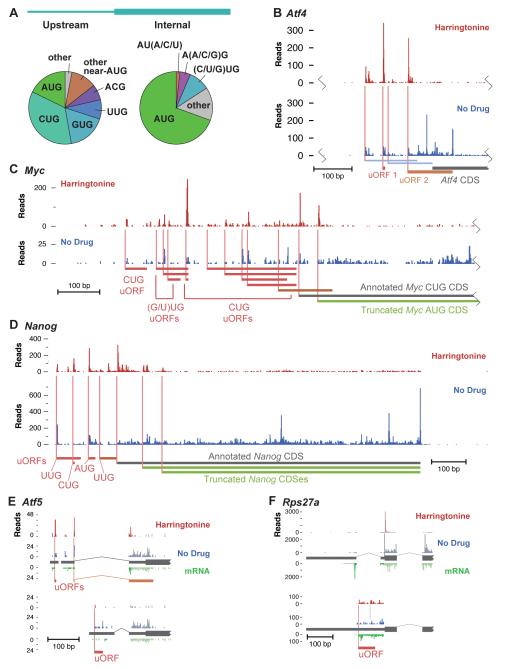
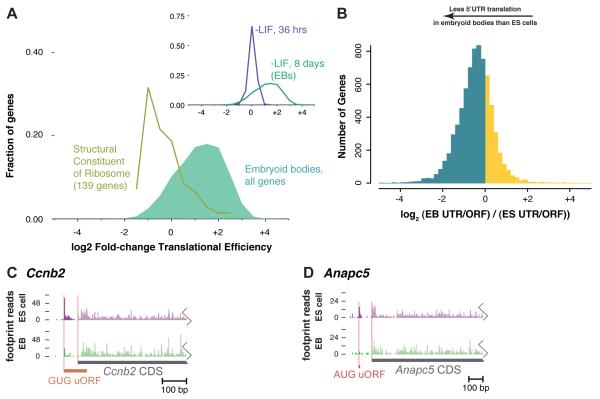
The ability to sequence genomes has far outstripped approaches for deciphering the information they encode. Here we present a suite of techniques, based on ribosome profiling (the deep sequencing of ribosome-protected mRNA fragments), to provide genome-wide maps of protein synthesis as well as a pulse-chase strategy for determining rates of translation elongation. We exploit the propensity of harringtonine to cause ribosomes to accumulate at sites of translation initiation together with a machine learning algorithm to define protein products systematically. Analysis of translation in mouse embryonic stem cells reveals thousands of strong pause sites and unannotated translation products. These include amino-terminal extensions and truncations and upstream open reading frames with regulatory potential, initiated at both AUG and non-AUG codons, whose translation changes after differentiation. We also define a class of short, polycistronic ribosome-associated coding RNAs (sprcRNAs) that encode small proteins. Our studies reveal an unanticipated complexity to mammalian proteomes.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–1136. - PubMed
-
- Atkins JF, Gesteland RF. Recoding: expansion of decoding rules enriches gene expression. New York, Springer: 2010.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
