

DNA methylation dynamics during in vivo differentiation of blood and skin stem cells
- PMID: 22841485
- PMCID: PMC3428428
- DOI: 10.1016/j.molcel.2012.06.019
DNA methylation dynamics during in vivo differentiation of blood and skin stem cells
Abstract
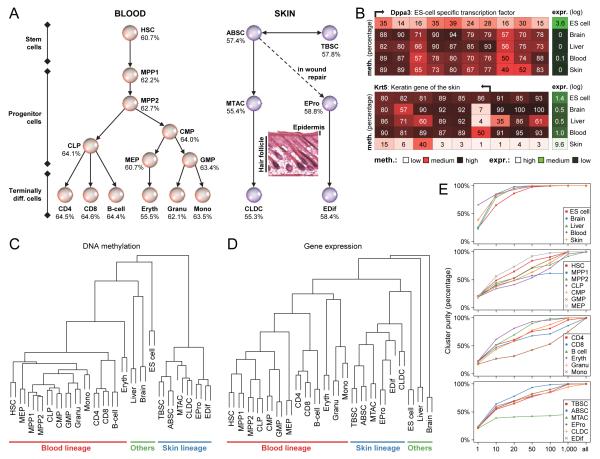
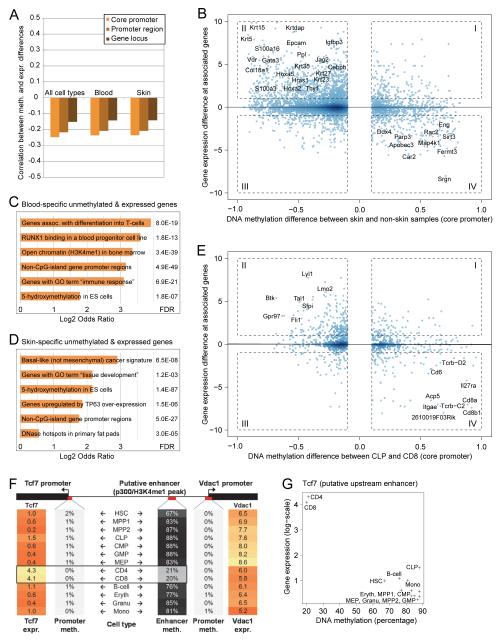
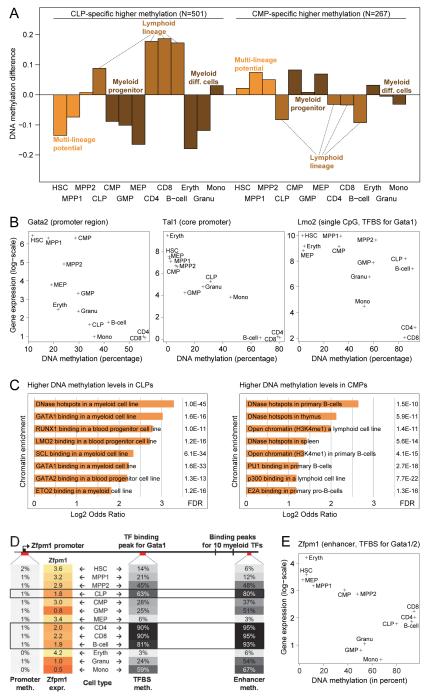
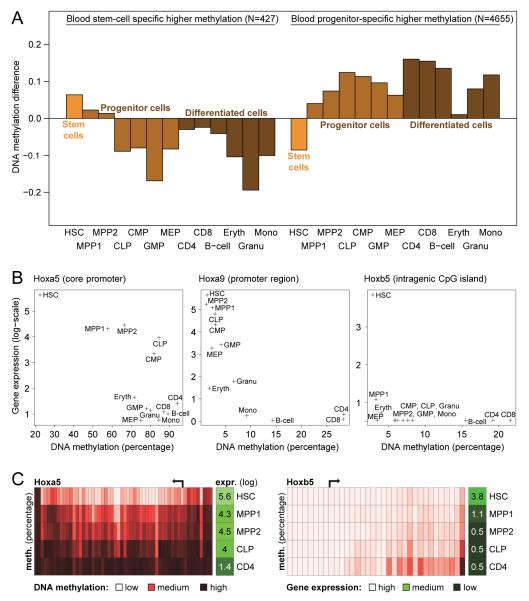
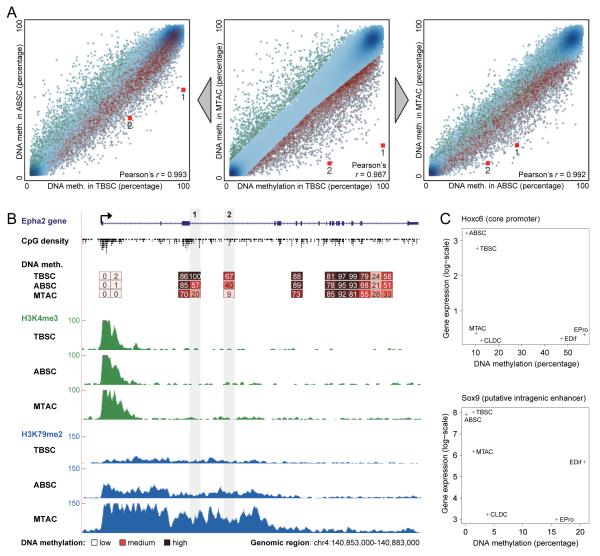
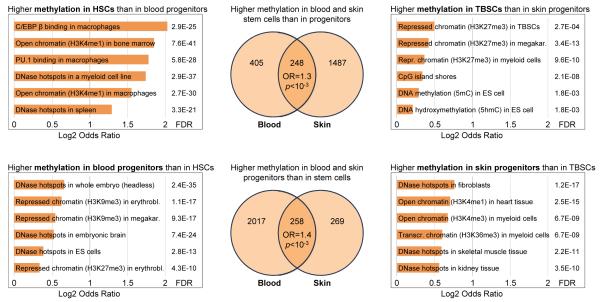
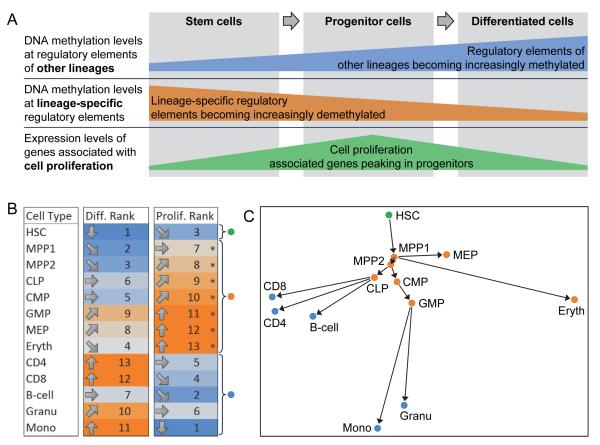
DNA methylation is a mechanism of epigenetic regulation that is common to all vertebrates. Functional studies underscore its relevance for tissue homeostasis, but the global dynamics of DNA methylation during in vivo differentiation remain underexplored. Here we report high-resolution DNA methylation maps of adult stem cell differentiation in mouse, focusing on 19 purified cell populations of the blood and skin lineages. DNA methylation changes were locus specific and relatively modest in magnitude. They frequently overlapped with lineage-associated transcription factors and their binding sites, suggesting that DNA methylation may protect cells from aberrant transcription factor activation. DNA methylation and gene expression provided complementary information, and combining the two enabled us to infer the cellular differentiation hierarchy of the blood lineage directly from genome-scale data. In summary, these results demonstrate that in vivo differentiation of adult stem cells is associated with small but informative changes in the genomic distribution of DNA methylation.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Adolfsson J, Mansson R, Buza-Vidas N, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
-
- Blanpain C, Lowry WE, Geoghegan A, et al. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–648. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
