

Transcriptome-wide association analyses reveal the impact of regulatory variants on rice panicle architecture and causal gene regulatory networks
- PMID: 37980346
- PMCID: PMC10657423
- DOI: 10.1038/s41467-023-43077-6
Transcriptome-wide association analyses reveal the impact of regulatory variants on rice panicle architecture and causal gene regulatory networks
Abstract
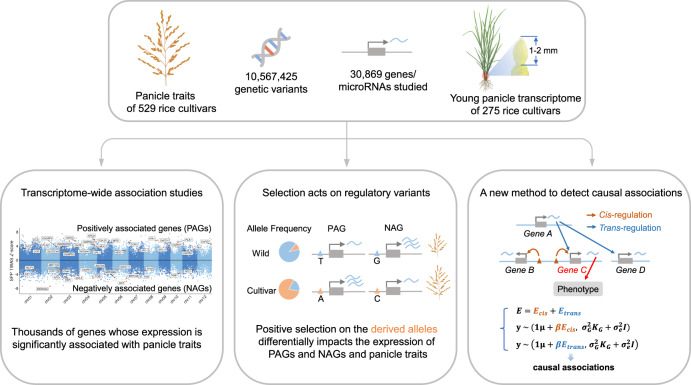
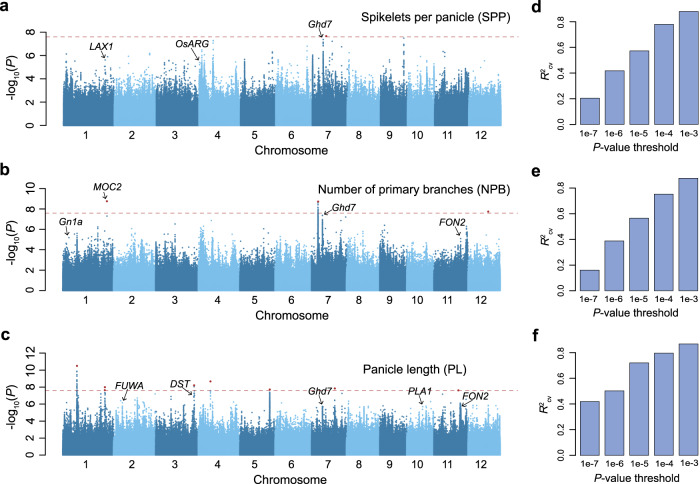
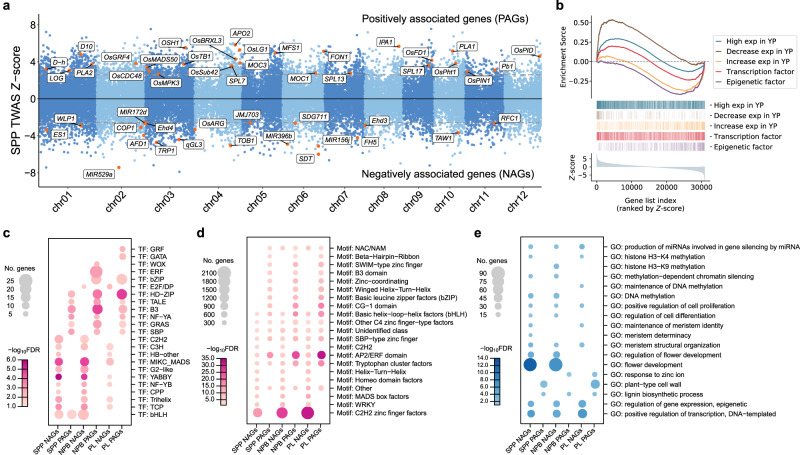
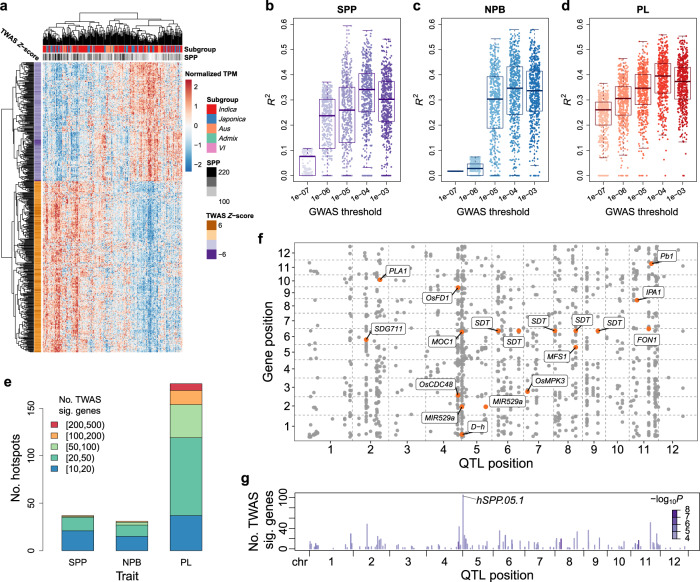
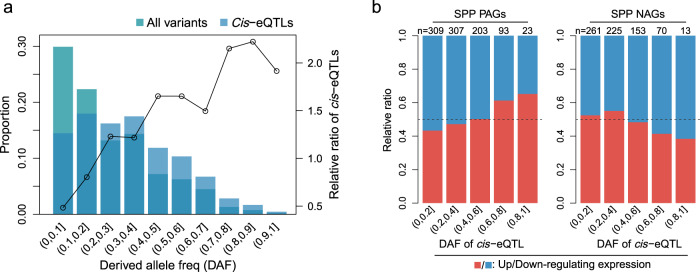
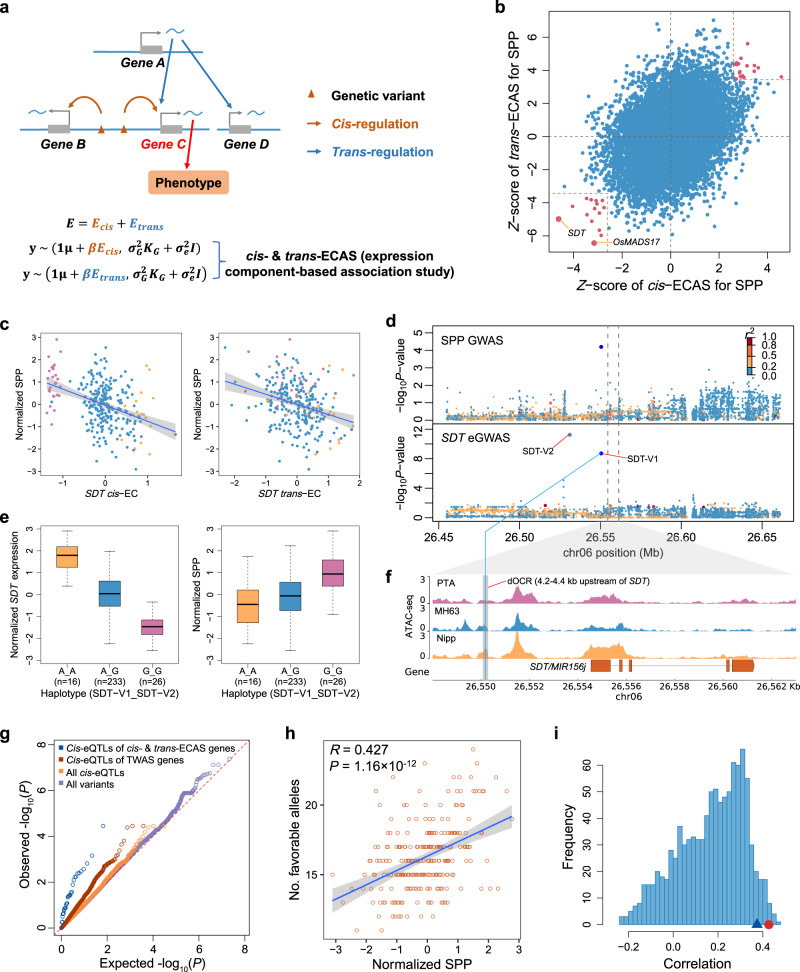
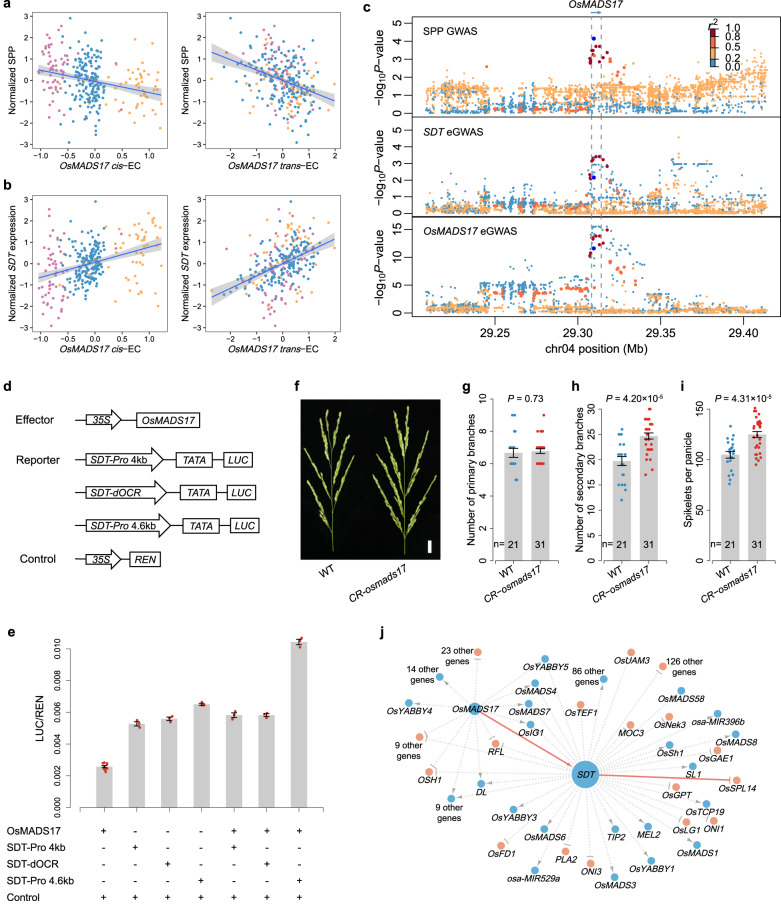
Panicle architecture is a key determinant of rice grain yield and is mainly determined at the 1-2 mm young panicle stage. Here, we investigated the transcriptome of the 1-2 mm young panicles from 275 rice varieties and identified thousands of genes whose expression levels were associated with panicle traits. Multimodel association studies suggested that many small-effect genetic loci determine spikelet per panicle (SPP) by regulating the expression of genes associated with panicle traits. We found that alleles at cis-expression quantitative trait loci of SPP-associated genes underwent positive selection, with a strong preference for alleles increasing SPP. We further developed a method that integrates the associations of cis- and trans-expression components of genes with traits to identify causal genes at even small-effect loci and construct regulatory networks. We identified 36 putative causal genes of SPP, including SDT (MIR156j) and OsMADS17, and inferred that OsMADS17 regulates SDT expression, which was experimentally validated. Our study reveals the impact of regulatory variants on rice panicle architecture and provides new insights into the gene regulatory networks of panicle traits.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Associated data
- BioProject/PRJCA012684
- Actions
- Actions
- Actions
- Actions
Grants and funding
- 31922065/National Natural Science Foundation of China (National Science Foundation of China)
- 31821005/National Natural Science Foundation of China (National Science Foundation of China)
- 32261143466/National Natural Science Foundation of China (National Science Foundation of China)
- 31630054/National Natural Science Foundation of China (National Science Foundation of China)
- 2023AFA043/Natural Science Foundation of Hubei Province (Hubei Provincial Natural Science Foundation)
LinkOut - more resources
Full Text Sources
Miscellaneous
