

High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3
- PMID: 24317040
- PMCID: PMC4009731
- DOI: 10.1038/ni.2784
High-density lipoprotein mediates anti-inflammatory reprogramming of macrophages via the transcriptional regulator ATF3
Abstract
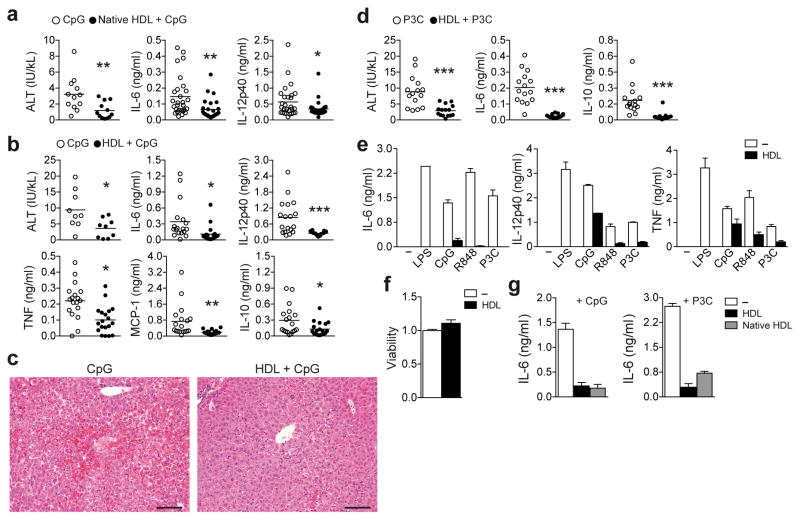
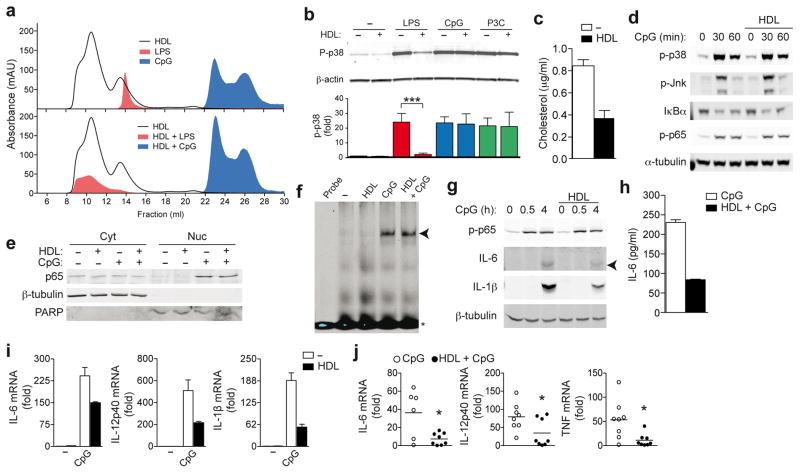
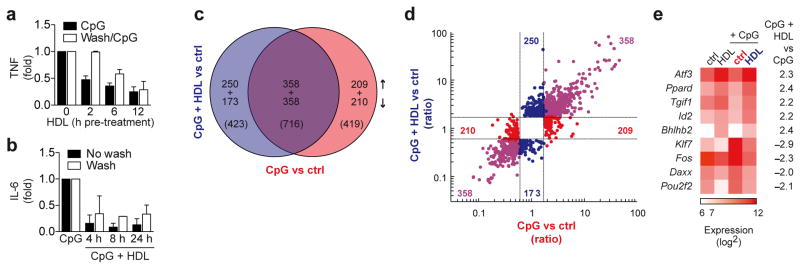
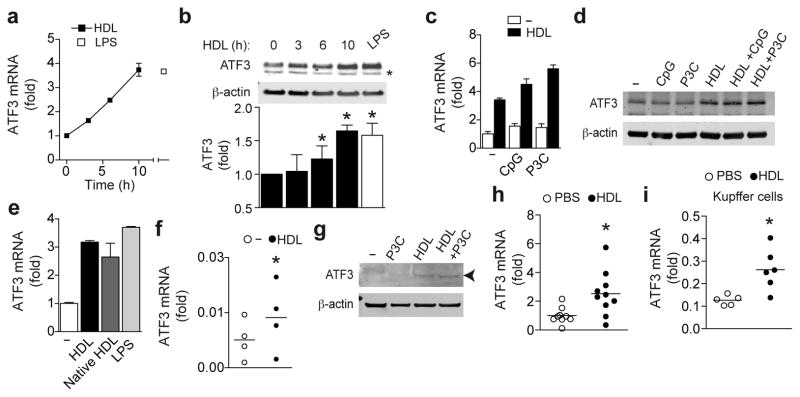
High-density lipoprotein (HDL) mediates reverse cholesterol transport and is known to be protective against atherosclerosis. In addition, HDL has potent anti-inflammatory properties that may be critical for protection against other inflammatory diseases. The molecular mechanisms of how HDL can modulate inflammation, particularly in immune cells such as macrophages, remain poorly understood. Here we identify the transcriptional regulator ATF3, as an HDL-inducible target gene in macrophages that downregulates the expression of Toll-like receptor (TLR)-induced proinflammatory cytokines. The protective effects of HDL against TLR-induced inflammation were fully dependent on ATF3 in vitro and in vivo. Our findings may explain the broad anti-inflammatory and metabolic actions of HDL and provide the basis for predicting the success of new HDL-based therapies.
Conflict of interest statement
S.D.W is a paid employee of CSL Behring.
Figures
Comment in
-
Old HDL learns a new (anti-inflammatory) trick.Nat Immunol. 2014 Feb;15(2):138-9. doi: 10.1038/ni.2798. Nat Immunol. 2014. PMID: 24448572 No abstract available.
-
High-density lipoproteins put out the fire.Cell Metab. 2014 Feb 4;19(2):175-6. doi: 10.1016/j.cmet.2014.01.009. Cell Metab. 2014. PMID: 24506861 Free PMC article.
-
Anti-inflammatory effects of high-density lipoprotein through activating transcription factor 3: benefit beyond cholesterol transport-dependent processes.Arterioscler Thromb Vasc Biol. 2014 Jun;34(6):e11-2. doi: 10.1161/ATVBAHA.114.303553. Epub 2014 Apr 17. Arterioscler Thromb Vasc Biol. 2014. PMID: 24743432 Free PMC article. No abstract available.
References
-
- Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. - PubMed
-
- Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. - PubMed
-
- Gilchrist M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. - PubMed
-
- Whitmore MM, et al. Negative regulation of TLR-signaling pathways by activating transcription factor-3. J Immunol. 2007;179:3622–3630. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
