

Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing
- PMID: 24179143
- PMCID: PMC3847781
- DOI: 10.1101/gr.161679.113
Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing
Abstract
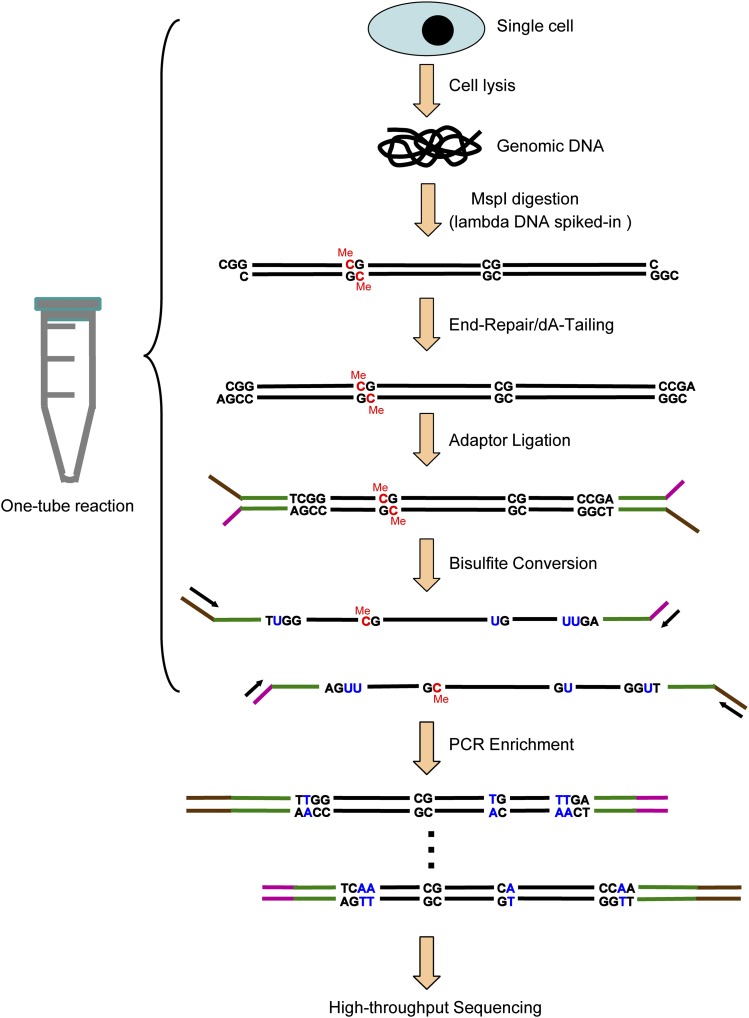
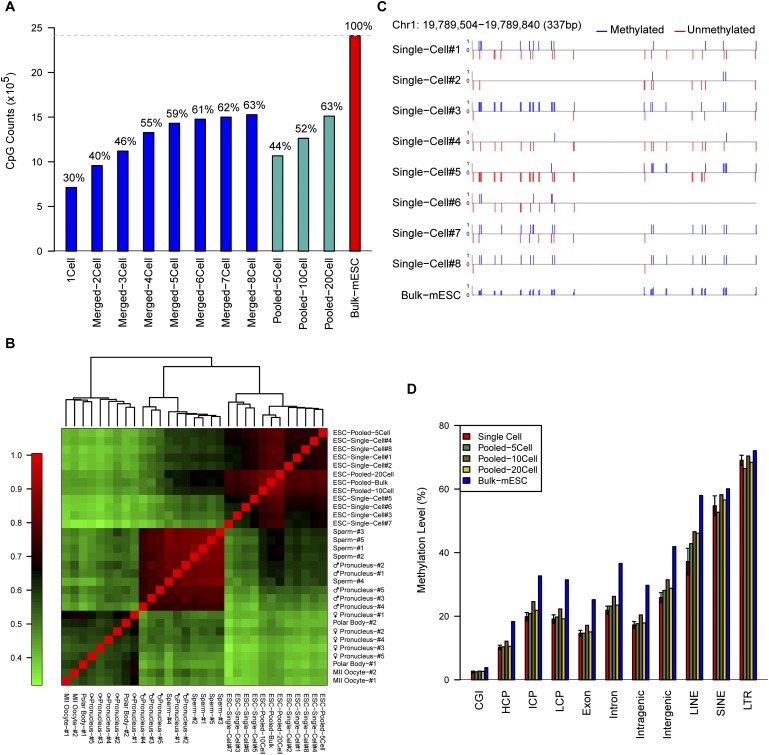
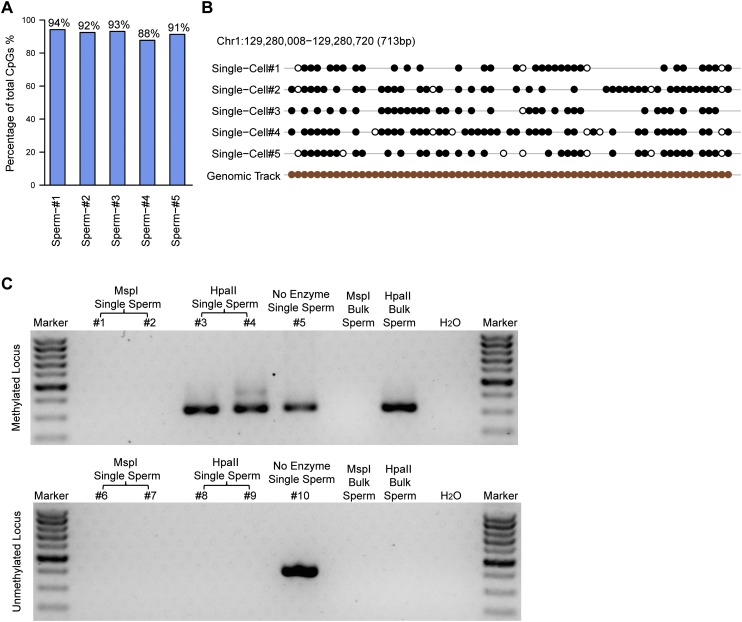
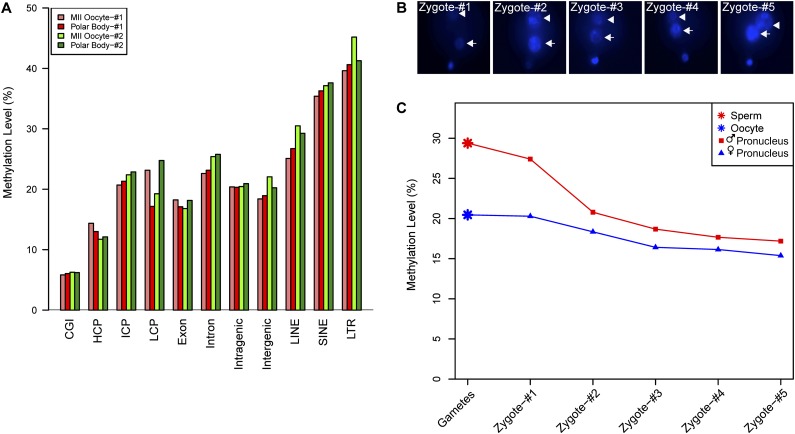
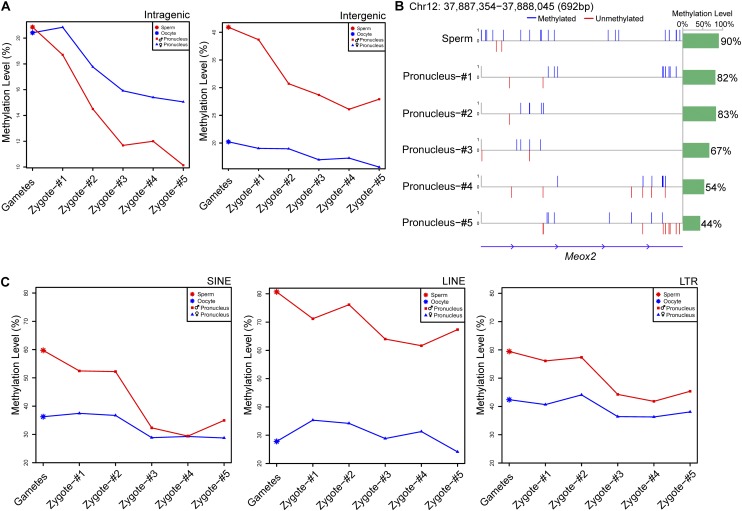
DNA methylation is crucial for a wide variety of biological processes, yet no technique suitable for the methylome analysis of DNA methylation at single-cell resolution is available. Here, we describe a methylome analysis technique that enables single-cell and single-base resolution DNA methylation analysis based on reduced representation bisulfite sequencing (scRRBS). The technique is highly sensitive and can detect the methylation status of up to 1.5 million CpG sites within the genome of an individual mouse embryonic stem cell (mESC). Moreover, we show that the technique can detect the methylation status of individual CpG sites in a haploid sperm cell in a digitized manner as either unmethylated or fully methylated. Furthermore, we show that the demethylation dynamics of maternal and paternal genomes after fertilization can be traced within the individual pronuclei of mouse zygotes. The demethylation process of the genic regions is faster than that of the intergenic regions in both male and female pronuclei. Our method paves the way for the exploration of the dynamic methylome landscapes of individual cells at single-base resolution during physiological processes such as embryonic development, or during pathological processes such as tumorigenesis.
Figures
References
-
- Bird A 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21 - PubMed
-
- Bock C 2012. Analysing and interpreting DNA methylation data. Nat Rev Genet 13: 705–719 - PubMed
-
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S 2012. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336: 934–937 - PubMed
Publication types
MeSH terms
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases