

The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts
- PMID: 24555846
- PMCID: PMC4053980
- DOI: 10.1186/gb-2014-15-2-r37
The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts
Abstract
Background: DNA methylation plays an essential role in the regulation of gene expression. While its presence near the transcription start site of a gene has been associated with reduced expression, the variation in methylation levels across individuals, its environmental or genetic causes, and its association with gene expression remain poorly understood.
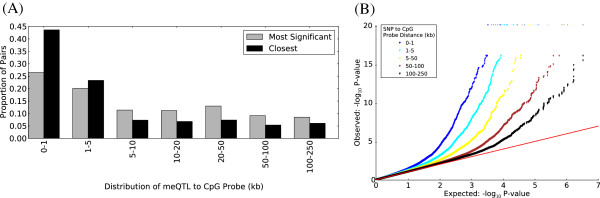
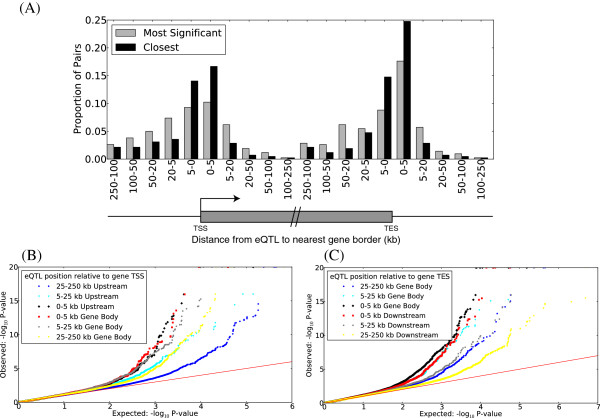
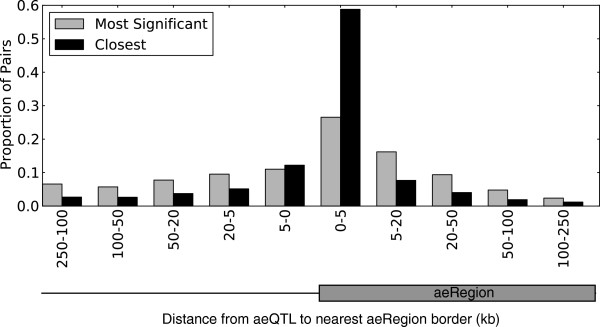
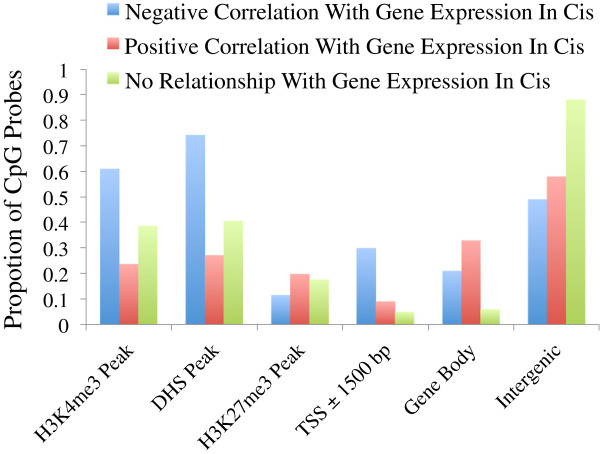
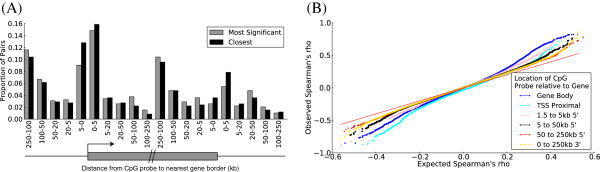
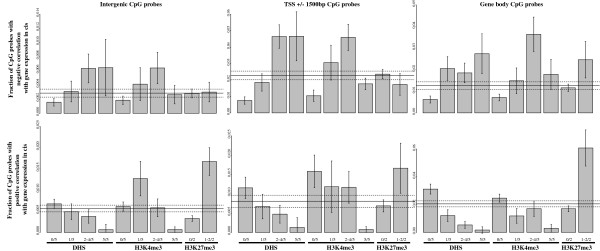
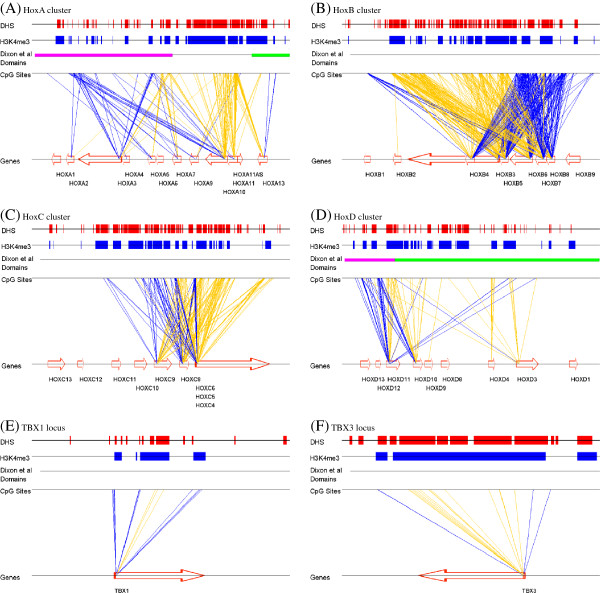
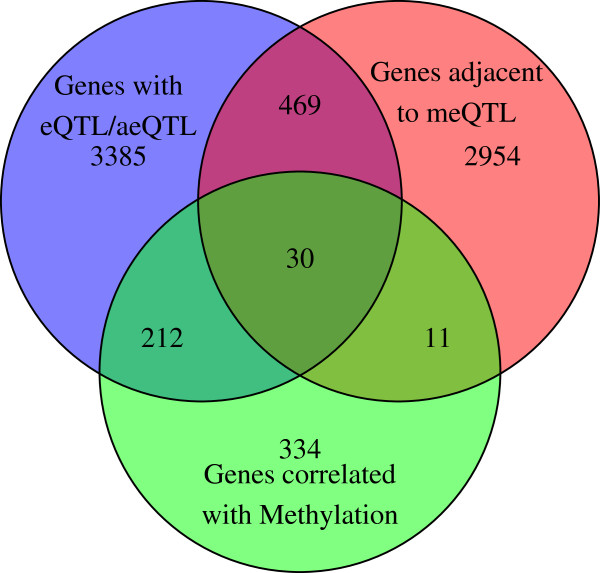
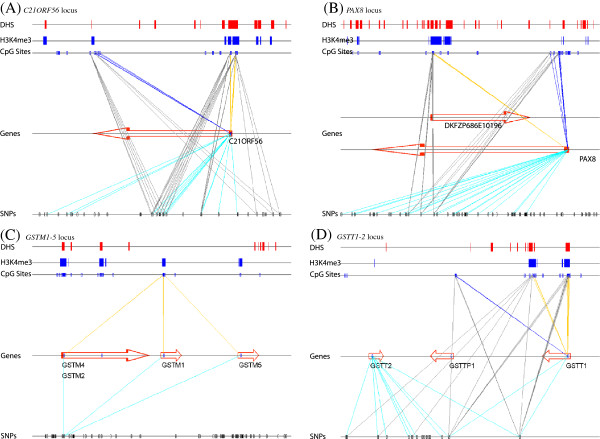
Results: We report the joint analysis of sequence variants, gene expression and DNA methylation in primary fibroblast samples derived from a set of 62 unrelated individuals. Approximately 2% of the most variable CpG sites are mappable in cis to sequence variation, usually within 5 kb. Via eQTL analysis with microarray data combined with mapping of allelic expression regions, we obtained a set of 2,770 regions mappable in cis to sequence variation. In 9.5% of these expressed regions, an associated SNP was also a methylation QTL. Methylation and gene expression are often correlated without direct discernible involvement of sequence variation, but not always in the expected direction of negative for promoter CpGs and positive for gene-body CpGs. Population-level correlation between methylation and expression is strongest in a subset of developmentally significant genes, including all four HOX clusters. The presence and sign of this correlation are best predicted using specific chromatin marks rather than position of the CpG site with respect to the gene.
Conclusions: Our results indicate a wide variety of relationships between gene expression, DNA methylation and sequence variation in untransformed adult human fibroblasts, with considerable involvement of chromatin features and some discernible involvement of sequence variation.
Figures
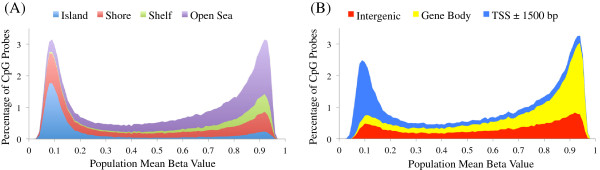
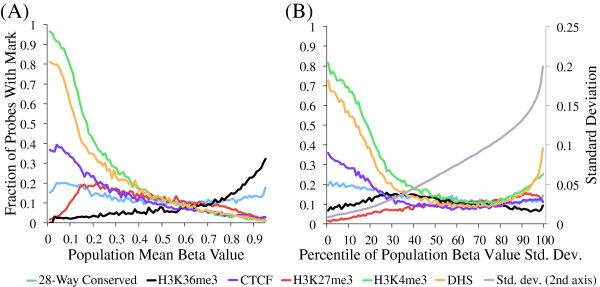
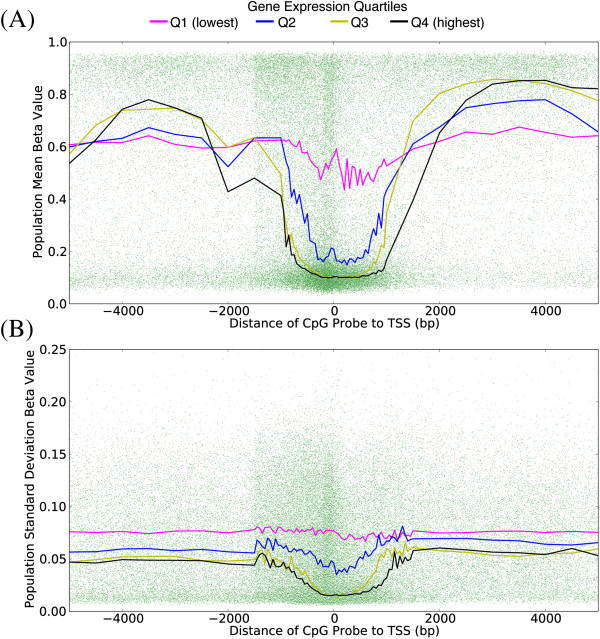
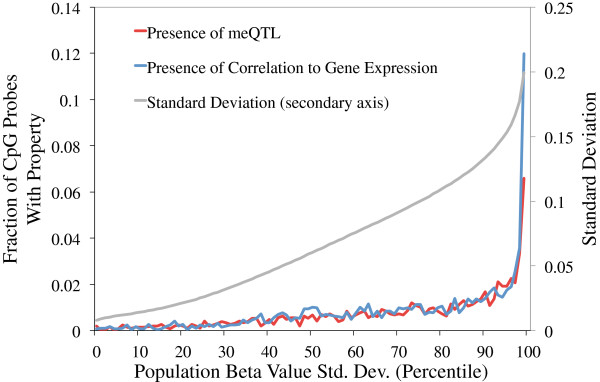
References
-
- Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
