

ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents
- PMID: 25100205
- PMCID: PMC4184920
- DOI: 10.1158/2159-8290.CD-14-0157
ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents
Abstract
Alkylating agents are a first-line therapy for the treatment of several aggressive cancers, including pediatric glioblastoma, a lethal tumor in children. Unfortunately, many tumors are resistant to this therapy. We sought to identify ways of sensitizing tumor cells to alkylating agents while leaving normal cells unharmed, increasing therapeutic response while minimizing toxicity. Using an siRNA screen targeting over 240 DNA damage response genes, we identified novel sensitizers to alkylating agents. In particular, the base excision repair (BER) pathway, including 3-methylpurine-DNA glycosylase (MPG), as well as ataxia telangiectasia mutated (ATM), were identified in our screen. Interestingly, we identified MPG as a direct novel substrate of ATM. ATM-mediated phosphorylation of MPG was required for enhanced MPG function. Importantly, combined inhibition or loss of MPG and ATM resulted in increased alkylating agent-induced cytotoxicity in vitro and prolonged survival in vivo. The discovery of the ATM-MPG axis will lead to improved treatment of alkylating agent-resistant tumors.
Significance: Inhibition of ATM and MPG-mediated BER cooperate to sensitize tumor cells to alkylating agents, impairing tumor growth in vitro and in vivo with no toxicity to normal cells, providing an ideal therapeutic window.
©2014 American Association for Cancer Research.
Conflict of interest statement
The authors have no conflicts or disclosures to declare
Figures

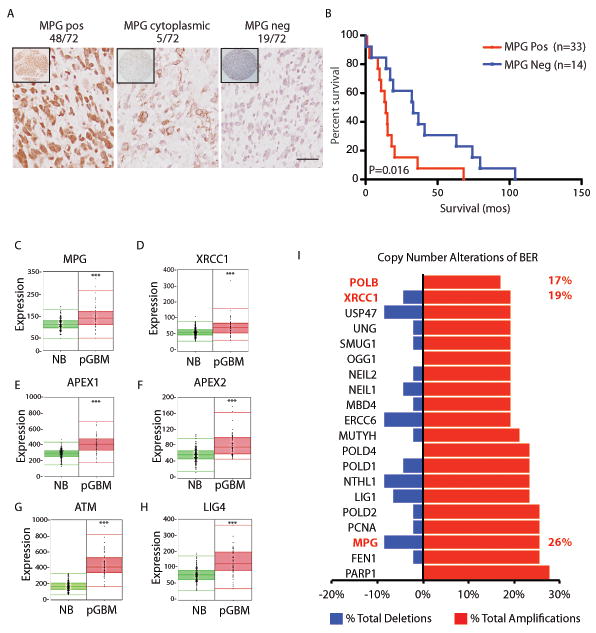

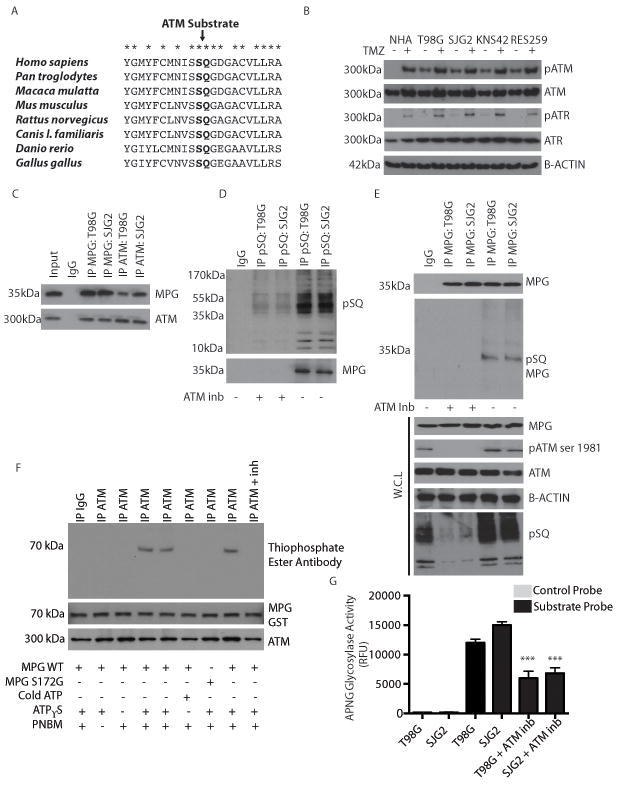
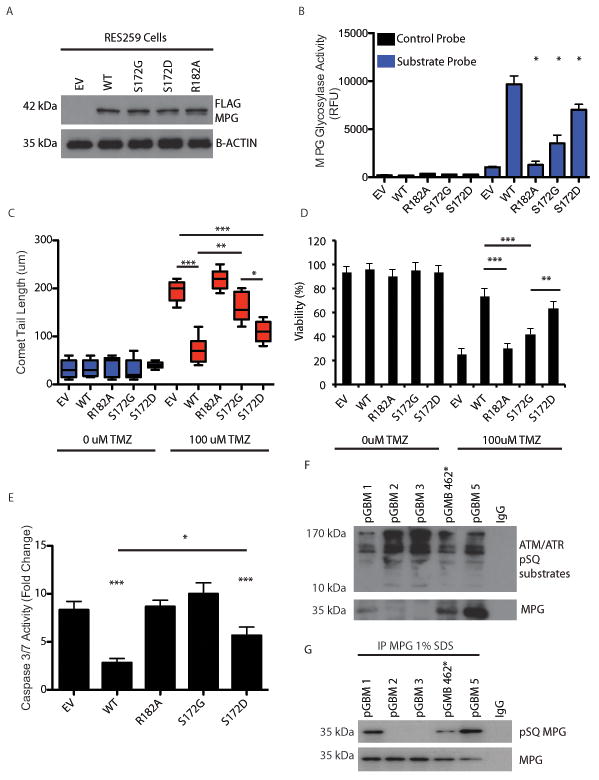

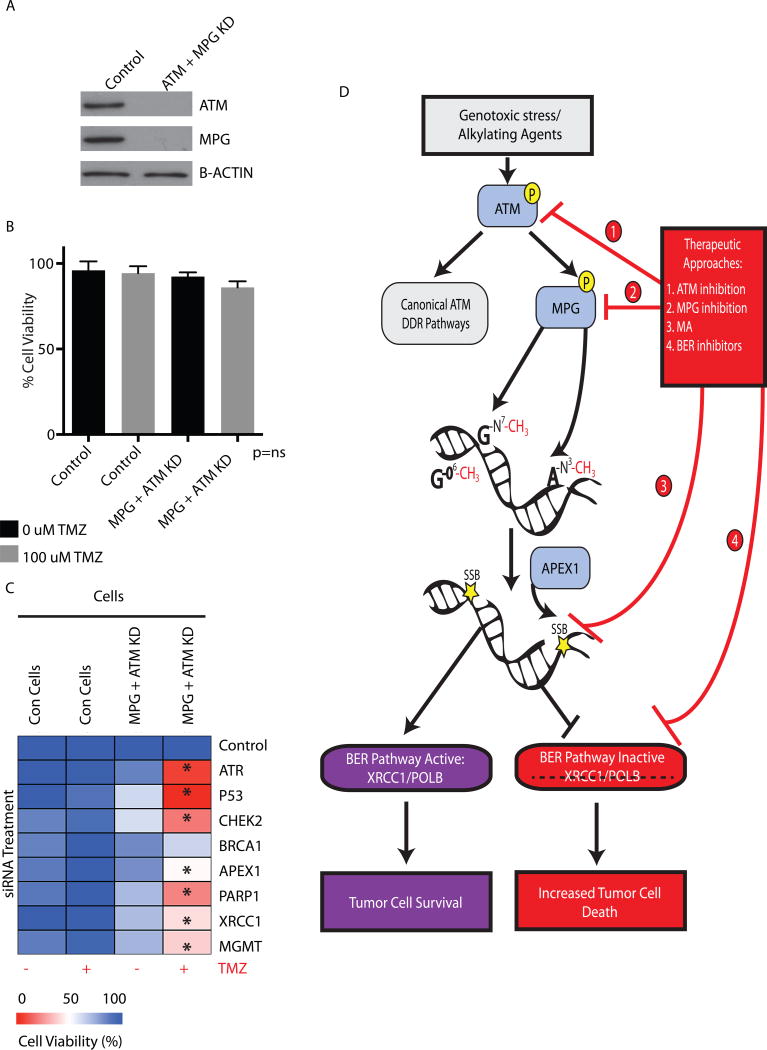
Comment in
-
Understanding and targeting alkylator resistance in glioblastoma.Cancer Discov. 2014 Oct;4(10):1120-2. doi: 10.1158/2159-8290.CD-14-0918. Cancer Discov. 2014. PMID: 25274683
References
-
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. - PubMed
-
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. - PubMed
-
- Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry. 1994;33:9045–51. - PubMed
-
- Hang B, Singer B, Margison GP, Elder RH. Targeted deletion of alkylpurine-DNA-N-glycosylase in mice eliminates repair of 1,N6-ethenoadenine and hypoxanthine but not of 3,N4-ethenocytosine or 8-oxoguanine. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:12869–74. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
