

NRAS mutation causes a human autoimmune lymphoproliferative syndrome
- PMID: 17517660
- PMCID: PMC1885609
- DOI: 10.1073/pnas.0702975104
NRAS mutation causes a human autoimmune lymphoproliferative syndrome
Abstract
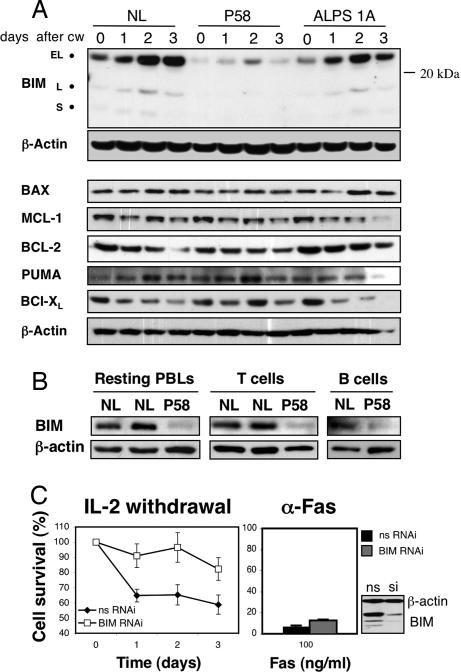
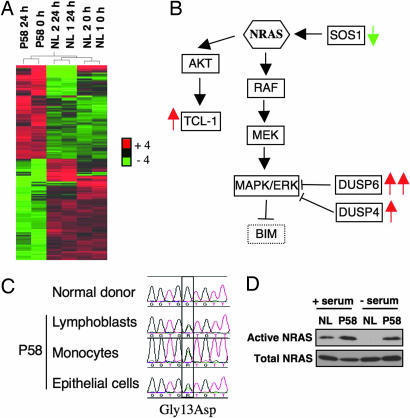
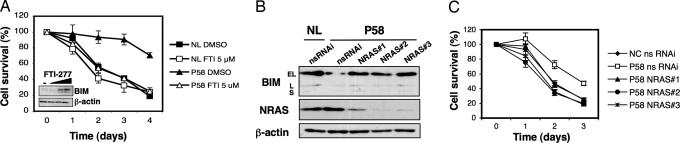
The p21 RAS subfamily of small GTPases, including KRAS, HRAS, and NRAS, regulates cell proliferation, cytoskeletal organization, and other signaling networks, and is the most frequent target of activating mutations in cancer. Activating germline mutations of KRAS and HRAS cause severe developmental abnormalities leading to Noonan, cardio-facial-cutaneous, and Costello syndrome, but activating germline mutations of NRAS have not been reported. Autoimmune lymphoproliferative syndrome (ALPS) is the most common genetic disease of lymphocyte apoptosis and causes autoimmunity as well as excessive lymphocyte accumulation, particularly of CD4(-), CD8(-) alphabeta T cells. Mutations in ALPS typically affect CD95 (Fas/APO-1)-mediated apoptosis, one of the extrinsic death pathways involving TNF receptor superfamily proteins, but certain ALPS individuals have no such mutations. We show here that the salient features of ALPS as well as a predisposition to hematological malignancies can be caused by a heterozygous germline Gly13Asp activating mutation of the NRAS oncogene that does not impair CD95-mediated apoptosis. The increase in active, GTP-bound NRAS augments RAF/MEK/ERK signaling, which markedly decreases the proapoptotic protein BIM and attenuates intrinsic, nonreceptor-mediated mitochondrial apoptosis. Thus, germline activating mutations in NRAS differ from other p21 Ras oncoproteins by causing selective immune abnormalities without general developmental defects. Our observations on the effects of NRAS activation indicate that RAS-inactivating drugs, such as farnesyltransferase inhibitors should be examined in human autoimmune and lymphocyte homeostasis disorders.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Malumbres M, Barbacid M. Nat Rev Cancer. 2003;3:459–465. - PubMed
-
- Barbacid M. Annu Rev Biochem. 1987;56:779–827. - PubMed
-
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, et al. Nat Genet. 2005;37:1038–1040. - PubMed
-
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, Step AL, Conger BA, Cruz MS, McCormick F, Paven KA. Science. 2006;3:1287–1290. - PubMed
-
- Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, et al. Nat Genet. 2006;38:331–336. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
